Нарушение системы гемостаза врожденные и приобретенные. Хирургические вмешательства, ожеги. Возникает чаще у детей
Система гемостаза - замечательное достижение эволюции, которое постоянно поддерживает баланс между двумя разнонаправленными процессами: максимально быстрым образованием сгустка (тромба) с целью предотвращения потери крови в ответ на повреждение сосуда и сохранением при этом жидкого агрегатного состояния крови в циркуляции. Решение этой трудной задачи обеспечивают сложные взаимодействия сосудистого эндотелия, плазменного коагуляционного каскада, антикоагулянтных механизмов, фибринолитической системы, тромбоцитов и лейкоцитов. Велико значение реологических характеристик движения крови по сосудам различного диаметра, особенно вязкости в системе микроциркуляции.
В медицине критических состояний нарушения гемостаза развиваются часто, степень их выраженности зависит от дерегулирующего действия на гомеостаз повреждающих факторов - травмы, инфекции, операции, медикаментов, а также компенсаторных возможностей сердечно-сосудистой и дыхательной систем. Расшифровка ведущей роли нарушений гемостаза, в частности синдрома диссеми-нированного внутрисосудистого свёртывания крови (ДВС-синдрома), в патогенезе большинства критических состояний, требующих интенсивной терапии, и появ-ление в арсенале врача эффективных целенаправленных средств его коррекции - отличительная черта современной медицинской практики.
Клинически нарушения гемостаза проявляются чаще кровотечениями, реже тромбозами, но нередко существует одновременное проявление патологической кровоточивости и микротромбирования.
Физиология нормального гемостаза
Повреждение стенки кровеносного сосуда вызывает немедленную вазокон- стрикцию как самого повреждённого сосуда, так и смежных капилляров и артери- ол, что приводит к начальному замедлению кровотока в зоне повреждения. Далее взаимодействие нескольких функциональных компонентов приводит к образова-нию первичной тромбоцитарной пробки (сгустка), быстро стабилизирующейся нитями фибрина. В нормальных условиях этот прокоагулянтный процесс огра-ничен по времени и месту, его контролируют те же функциональные компоненты. К ним относят тромбоциты, плазменный коагуляционный каскад, естественные антикоагулянты, систему фибринолиза и эндотелиальные клетки.
Тромбоциты при повреждении эндотелия вступают в контакт с субэндотелиаль- ным слоем коллагена, происходит адгезия (прилипание) тромбоцитов к коллагену, образуются тромбоцитарные гранулы с высвобождением серотонина, лизосомаль- ных ферментов, фибриногена и IV тромбоцитарного фактора, стимулируется синтез простагландина. Активированные тромбоциты агрегируют, образуя первичную тромбоцитарную пробку (первичный гемостаз). Количественные и качественные патологические изменения тромбоцитов проявляются кровоточивостью и кровоизлияниями. Время кровотечения - оптимальный скрининговый тест для оценки функциональной состоятельности тромбоцитов.
Коагуляционный каскад отвечает за формирование стабильного фибринового тромба. Факторы, составляющие его, перечислены в табл. 2-10.
Таблица 2-10. Факторы свёртывания Фактор 1 Фибриноген Фактор II Протромбин Фактор III Тканевой тромбопластин (тканевой фактор) Фактор IV Кальций Фактор V Акцелерин Фактор VII Проконвертин Фактор VIII Антигемофильный фактор Фактор IX Кристмас-фактор Фактор X Фактор Стюарта Фактор XI Плазменный предшественник тромбопластина Фактор XII Фактор Хагемана Фактор XIII Фибринстабилизирующий фактор
Синтез большинства факторов свёртывания происходит в печени. Фактор VIII, кроме печени, частично синтезируют мегакариоциты и эндотелиальные клетки. Факторы II, VII, IX и X витамин К зависимы, т.е. для их синтеза гепатоцитами необходим витамин К. Схема коагуляционного каскада свёртывания представлена на рис. 2-8.
Началом коагуляционного каскада служит взаимодействие фактора VII с тканевым фактором (III) в зоне повреждения (внешний путь свёртывания). Образующийся комплекс активированного фактора VII с тканевым фактором активирует факторы IX и X, что приводит к образованию тромбина. Тромбин превращает фибриноген в фибрин, активируя фактор XIII и тромбоциты. Действие тромбина находится под контролем естественного антикоагулянта - активированного протеина С. Альтернатива внешнему пути свёртывания - внутренний, начинающийся с активации фактора XII коллагеном вследствие контакта крови с чужеродной поверхностью. Фактор ХНа активирует фактор Х1а, далее внешний и внутренний пути свёртывания идентичны.
Внутренний путь Внешний путь
(фактор контакта) (тканевые повреждения)
РШ
РХа
РУа
РХШа
РХШ
Рис. 2-8. Схема коагуляционного каскада.
Скрининговые тесты - протромбиновое время, активированное частичное тромбопластиновое время (АЧТВ) и тромбиновое время - позволяют судить о состоянии звеньев коагуляционного гемостаза. Врождённые или приобретённые дефекты коагуляционного каскада служат причиной геморрагий в мозг, суставы, мягкие ткани и мышцы, желудочно-кишечных кровотечений.
Естественные антикоагулянты представлены двумя важнейшими факторами - антитромбином III и витамин К зависимыми протеинами С и 5. Антитромбин III ингибирует тромбин и фактор Ха, участвует в инактивации факторов 1Ха, Х1а, ХИа. Взаимодействуя с гепарином, антитромбин III значительно усиливает его анти- коагулянтный эффект. Активированный протеин С в соединении с протеином 5 оказывает антикоагулянтное действие на ферменты, ингибирующие факторы Уа и УШа (важные кофакторы прокоагулянтного процесса). Кроме этого, протеин С усиливает фибринолиз, ингибируя тканевой активатор плазминогена, облегчая переход плазминогена в плазмин. Важную роль играет протеин С в патогенезе
ДВС-синдрома. Дефицит антитромбина III, протеина С и протеина 5 (чаще бывает приобретённый, чем врождённый) сопровождается высокой частотой тромботических осложнений.
Фибринолиз (подобно коагуляции) - нормальный ответ на повреждение сосудистой стенки. Тканевой активатор плазминогена (1-РА), образуемый эндотелиальными клетками в ответ на их повреждение или стимуляцию тромбином, преобразует плазминоген в плазмин. Плазмин разрушает фибрин и фибриноген, образуя различные продукты деградации фибрина. Одновременно в плазме циркулирует в неактивной форме ингибитор активатора плазминогена (РА1-1, образуемый гепа- тоцитами и эндотелиальными клетками, и РА1-2, образуемый в плаценте и макрофагах). Их физиологическое значение заключается в контролировании фибрино- лиза и предупреждении его перехода в патологическую стадию. Терапевтические ингибиторы фибринолиза - транексамовая кислота и аминокапроновая кислота. Напротив, стрептокиназа - фибринолитический препарат, повышающий образо-вание плазмина.
Эндотелиальные клетки в нормальных условиях ответственны за антитромбо- тическое взаимодействие между кровью и тканями, поддерживая жидкое состояние крови. Они продуцируют такие антикоагулянты, как гликозаминогликаны, гепаринсульфаты, тромбомодулин (активируют антитромбин III и протеин С), оксид азота и простагландин (ингибируют агрегацию тромбоцитов и способствуют вазодилатации), тканевой активатор плазминогена, инициирующий фибринолиз. Но эндотелиальные клетки способны в ответ на стимуляцию бактериальными эндотоксинами продуцировать на своей поверхности фактор Виллебранда и тканевой фактор, которые запускают коагуляционный каскад. Эти их свойства необходимо учитывать при проведении терапии нарушений гемостаза.
Лабораторный скрининг нарушений системы гемостаза
Тесты для оценки первичного гемостаза, выполняемые экстренно в условиях проведения интенсивной терапии, - это определение времени кровотечения и количества тромбоцитов. Агрегацию и адгезию тромбоцитов, содержание фактора Виллебранда исследуют в специализированных коагулологических лабораториях.
Процесс коагуляционного гемостаза можно оценить такими тестами, как АЧТВ, протромбиновое время и тромбиновое время, которые дают суммарную оценку некоторых факторов свёртывания крови. Раздельное определение факторов свёр-тывания возможно только в специализированных лабораториях.
Скрининг противосвёртывающей системы может быть осуществлён определением концентрации антитромбина III; другие тесты ингибирования свёртывания занимают длительное время, их проводят в специализированной лаборатории.
О состоянии фибринолитической системы можно судить, исследуя количество продуктов деградации фибриногена, мономеров фибрина, Б-димеров, а также с помощью специфических исследований содержания в плазме активаторов и инги-биторов фибринолиза.
Нормальные значения коагуляционных показателей приведены в табл. 2-11.
Таблица 2-11. Показатели свёртывания крови Тест Норма АЧТВ 27,4-40,3 с Протромбиновое время 12,3-16,1 с Тромбиновое время Контроль ±3 с Фибриноген 1,7-3,1 г/л Р-димеры Окончание табл. 2-11 Факторы свёртывания II, V, VII, VIII, IX, X, XI, XII 50-150% Антитромбин III 80-120% Активированный протеин С 73-121% Протеин 3: общий 55-125% свободный 21-53% Фактор Виллебранда (антиген) 50-150%
Время кровотечения соответствует времени, которое необходимо для образо-вания тромбоцитарной пробки. Оно может быть удлинено при тромбоцитопении, дисфункции тромбоцитов и болезни Виллебранда, но никогда не удлиняется при коагулологических нарушениях (например, при гемофилии).
Подсчёт количества тромбоцитов необходимо включать в обязательный лабораторный скрининг у пациентов отделений интенсивной терапии. Следует помнить, что количество тромбоцитов может быть снижено в первые дни менструации.
АЧТВ отражает суммарное содержание всех факторов внутреннего пути свёр-тывания - от активации фактора XII до образования растворимого фибрина, кроме факторов VII и XIII. Удлинение АЧТВ отражает повышенную тенденцию к кровоточивости и может указывать на дефицит одного или нескольких факторов внутреннего звена гемостаза (например, VIII при гемофилии или болезни Виллебранда). Удлиняет АЧТВ терапия гепарином натрия или варфарином. Нарушения первичного гемостаза АЧТВ не отражает.
Протромбиновое время характеризует процесс формирования сгустка. Оно определяет сумму факторов свёртывания I, II, V, VII и X, составляющих внешний путь, и удлиняется при дефиците одного или нескольких из них. Этот показатель нередко используют для контроля терапии непрямыми антикоагулянтами (варфарином) - антагонистами витамина К, которые соответственно не действуют на фибриноген и фактор V. Более информативно в этих целях исследование протромбинового комплекса, который определяет только сумму факторов II, VII и X (для синтеза этих факторов необходим витамин К). С целью сравнения результатов исследований различных лабораторий, пользующихся разными тест-системами, показатели протромбинового времени и протромбинового комплекса пересчиты-вают в международное нормализованное отношение (МНО). У здоровых лиц МНО составляет около 1. При лечении варфарином, дефиците витамина К, печёночной недостаточности или изолированном дефиците одного из факторов (обычно VII) значения МНО должны быть между 2 и 3.
Тромбиновое время тестирует конечный этап тромбообразования. Его удлинение может говорить о фибриногенопении менее 1 г/л или о дисфибриногене- мии. Терапия гепарином натрия также удлиняет тромбиновое время.
Низкое содержание фибриногена может быть следствием сниженной его продукции или увеличенного потребления. Высокое содержание фибриногена - показатель острого воспалительного состояния, особенно печени, где происходит его синтез. Повышение концентрации продуктов лизиса фибриногена (продукты деградации фибрина, Б-димер) при одновременном снижении его количества говорит о развитии ДВС-синдрома.
Дефицит антитромбина III (наиболее мощного естественного протеолитиче- ского плазменного антикоагулянта) часто служит проявлением острой массивной кровопотери, неадекватно восполненной [без переливания свежезамороженной плазмы (СЗП)], или ДВС-синдрома, сопровождающего сепсис. Значительно реже уменьшение количества антитромбина III возникает как проявление наследствен-ного заболевания аутосомно-доминантного характера. Использование скрининго-вых тестов для дифференциальной диагностики приобретённых геморрагических синдромов или заболеваний, основным клиническим проявлением которых слу-жит патология системы гемостаза, показано в табл. 2-12.
Таблица 2-12. Исследование показателей гемостаза при некоторых заболеваниях и синдромах Тест Гемофилия Болезнь
Виллебранда Острый
двс-
синдром болезни
печени Гепарин
натрия Варфарин Тромбоциты Норма Норма Снижено Норма Норма Норма Фибриноген Норма Норма Снижен Снижен Норма Норма Протромбиновое
время Норма Норма Удлинено Удлинено Норма Удлинено АЧТВ Удлинено Норма или удлинено Норма или удлинено Норма или удлинено Удлинено Удлинено Тромбиновое время Норма Норма Удлинено Норма или удлинено Удлинено Норма
Однако перечисленные классические тесты мониторинга гемостаза, к сожалению, не дают интегральной, цельной картины его состояния, отражающей взаимо-действие многочисленных факторов. В интенсивной терапии зачастую необходимо знать о состоянии системы свёртывания крови у пациента в момент исследования как с целью выбора медикаментозных или трансфузионных средств коррекции, так и с целью оценки правильности терапии, проводимой для коррекции нарушений гемостаза. Метод тромбоэластографии позволяет быстро и надёжно получать данные, характеризующие как общее состояние гемостаза, так и состояние отдельных его звеньев при их патологических изменениях.
Метод тромбоэластографии основан на измерении физических параметров сгустка (тромба) в процессе его образования. С момента начала свёртывания и до его окончания и последующего лизиса с помощью электромеханического преоб-разователя регистрируют изменение плотности сгустка, которое в современных моделях тромбоэластографа передаётся на дисплей компьютера. Различают сле-дующие основные параметры тромбоэластографии (рис. 2-9):
К (мин) - время от начала свёртывания до образования первых волокон фибрина;
К (мин) - время изменения амплитуды свёртывания, его нарастания или замедления;
К+К - коррелирует со временем свёртывания крови, равным в норме 6-8 мин;
угол а - отражает скорость свёртывания крови, процесс полимеризации фибрина;
МА (мм) - максимальная амплитуда кривой, характеризующая силу и жёсткость образовавшегося сгустка, которые зависят преимущественно от функции и количества тромбоцитов и, в меньшей степени, от концентрации фибрина в крови;
ЕРЬ, ЬУЗО (%) - оценка степени фибринолиза.
Как видно из рис. 2-9, при гиперкоагуляционном синдроме временные показатели КиК укорачиваются, угол а и МА увеличиваются. Напротив, при гипокоагу- ляционном статусе КиК увеличиваются, угол а и МА уменьшаются. Кроме общей характеристики состояния гемостаза, по данным тромбоэластографии можно также судить и о нарушениях его отдельных звеньев. Так, при передозировке гепа-рина натрия будут удлинены и снижены все показатели тромбоэластограммы - К, К, а, МА; при тромбоцитопении К останется в пределах нормы, но К будет удлинено при сниженной МА; при усиленном фибринолизе # нормальное, но МА резко и длительно снижена ЕРЬ и ЬУЗО; при тромбоцитопатии и дисфункции тромбоцитов
Начало
Рис. 2-9. Варианты тромбоэластограммы: А - норма; Б - гиперкоагуляция; В - гипокоагуляция.
К удлинено, угол а и МА уменьшены. Возможность неоднократного выполнения тромбоэластографии, графическая её запись, минимальные затраты времени на исследование при очень небольшом объёме крови, необходимом для его выполне-ния, делают тромбоэластограф одним из обязательных атрибутов лабораторного оборудования экспресс-лаборатории отделений интенсивной терапии.
ИНТЕНСИВНАЯ ТЕРАПИЯ ОТДЕЛЬНЫХ ФОРМ НАРУШЕНИЙ СИСТЕМЫ ГЕМОСТАЗА
В медицине критических состояний полное коагулологическое исследование нередко не может быть выполнено из-за тяжести состояния больного, дефицита времени и экстренности оперативного вмешательства. Следует отметить, что подозрение на наличие кровотечения вследствие системного нарушения гемостаза в подавляющем большинстве случаев может быть исключено, если у пациента гема- токрит >30%, количество тромбоцитов >100х109/л, концентрация фибриногена >1,5 г/л и при этом АЧТВ и протромбиновое время не более чем на 3-5 с превышают верхние границы нормальных значений. При таких показателях возможно безопасное выполнение инвазивных процедур (катетеризации центральной вены или пункции артерии). В то же время кровотечение при этих показателях может указывать на дефект хирургического локального гемостаза (обычно после операции) или на развитие острого ДВС-синдрома.
Наиболее часто встречающиеся системные нарушения гемостаза, требующие интенсивной терапии, приведены в табл. 2-13.
Таблица 2-13. Нарушения гемостаза при критических состояниях Диагноз Патология гемостаза Болезни печени Снижение содержания всех факторов свертывания, кроме VII и фактора Виллебранда Уменьшение концентрации протеина С и 5 Уменьшение количества тромбоцитов и их дисфункция ДВС-синдром Дисфибриногенемия опн Дисфункция тромбоцитов Уменьшение концентрации антитромбина III ДВС-синдром Аортокоронарное шунтирование с использованием АИК Дисфункция тромбоцитов Уменьшение количества тромбоцитов Уменьшение концентрации фибриногена Уменьшение содержания факторов II, V, VII, X, XI ДВС-синдром ЧМТ, краш-синдром Повышение концентрации продуктов деградации фибрина ДВС-синдром Массивные трансфузии Уменьшение содержания факторов V и VII ДВС-синдром Варфарин Уменьшение содержания факторов II, VII, IX, X Уменьшение концентрации протеина С и 5 Гепарин натрия Тромбоцитопения Уменьшение содержания фактора Ха Тромболитическая
терапия Повышение концентрации продуктов деградации фибрина, плазмина Уменьшение содержания фибриногена ДВС-синдром Уменьшение содержания фибриногена Уменьшение количества тромбоцитов Уменьшение содержания протеина С и 5 Уменьшение концентрации антитромбина III Уменьшение содержания факторов V, VIII, IX, XI Повышение концентрации продуктов деградации фибрина
Минимально достаточное содержание плазменных факторов свёртывания в русле крови, необходимое для обеспечения гемостаза во время проведения опера-тивных вмешательств, иллюстрирует табл. 2-14.
Минимально достаточное содержание тромбоцитов составляет 80-100х109/л.
Таблица 2-14. Минимальное содержание факторов свёртывания, необходимое для хирургического гемостаза Фактор Гемостатический уровень, % нормы Фибриноген 50-100 Протромбин 40-50 Фактор V 10-30 Фактор VII 10-20 Фактор VIII 30-70 Фактор Виллебранда 20-50
Окончание табл. 2-14 Фактор IX 20-60 Фактор X 10-20 Фактор XI 20-80 Фактор XIII 10
Нарушения первичного гемостаза
К геморрагическим диатезам, вызванным поражением первичного гемостаза, относят тромбоцитопению (см. главу 9), болезнь Виллебранда и нарушения функции тромбоцитов. Характерные симптомы - геморрагические высыпания на коже или слизистых оболочках, появление синяков, петехий при минимальном воздействии. Интенсивная терапия необходима при локальных кровотечениях, особенно часто при носовом и меноррагии.
Тромбоцитопению, обусловленную поражением костного мозга (апластиче- ская анемия, гемобластозы, метастазы рака), регистрируют наиболее часто. Реже в практике врача интенсивной терапии встречаются больные, геморрагический тромбоцитопенический диатез которых обусловлен врождёнными причинами (синдром Вискотта-Олдрича, аномалия Мая-Хегглина) или радиационным поражением, дефицитом железа, витамина В12, хроническим алкоголизмом.
Идиопатическая тромбоцитопеническая пурпура - наиболее распространённая форма аутоиммунной тромбоцитопении, для которой характерно укорочение жизни тромбоцитов вследствие их повышенного потребления. Функция тромбоцитов не страдает. При появлении кровотечений или для их профилактики назначают глюкокортикоиды. Эффективно применение высоких доз внутривенного иммуноглобулина. Нередко, особенно при появлении осложнений глюко- кортикоидной терапии (синдром Иценко-Кушинга, гипергликемия) или рецидивов кровотечения, прибегают к спленэктомии. В день операции и в ближайшем послеоперационном периоде глюкокортикоидную терапию продолжают, отмена её должна быть постепенной (снижение дозы на 2,5-5 мг преднизолона в сутки). Трансфузии тромбоцитов не считают средством выбора, кроме крайне редких случаев (неэффективность консервативной терапии и необходимость предупреждения повышенной кровоточивости во время операции).
Иммунный конфликт служит также основой гепарин-индуцированной тромбоцитопении. Организм пациента в ответ на введение гепарина натрия начинает образовывать антитела 1§С, которые связываются с Рс-рецепторами тромбоцитов, способствуя укорочению их жизни. Частота встречаемости такого конфликта -
5% лиц, получающих гепарин натрия. Тромбоцитопения развивается медленно, локальные кровотечения редки, в переливании тромбоцитов нет необходимости. При отмене гепарина натрия количество тромбоцитов восстанавливается в течение 2-5 дней.
Многие медикаменты, применяемые в интенсивной терапии, могут вызывать нарушения тромбоцитарной функции, но они, как правило, клинически незначимы. Лишь некоторые могут вызвать кровотечение.
Ацетилсалициловая кислота блокирует агрегацию тромбоцитов, ингибирование продолжается на протяжении всей жизни тромбоцитов в циркуляции (7-10 дней), следовательно, даже однократный приём ацетилсалициловой кислоты может на достаточно длительный срок блокировать важнейшую функцию первичного гемостаза. При необходимости выполнения операций в такой ситуации показано назначение десмопрессина, который временно (на три часа) устраняет удлинение времени кровотечения. Повторное назначение десмопрессина продляет его действие.
НПВП блокируют синтез тромбоксана А2 - важного медиатора агрегации тромбоцитов. Их действие также может быть нейтрализовано назначением дес- мопрессина.
Растворы декстранов снижают концентрацию в плазме фактора VIII, фактора Виллебранда, ингибируют функцию тромбоцитов, удлиняя время кровотечения. Несмотря на своё хорошее объёмозамещающее действие, переливание декстранов при острой массивной кровопотере или при наличии исходно напряжённого гемостаза (гемофилии, болезнях системы крови, болезнях печени и др.) в настоящее время должно быть сведено к минимуму.
Клопидогрел, тиклопидин, а также современные ингибиторы тромбоцитарно- го рецептора СРПЬ/Ша (абциксимаб), применяемые у больных с ишемической болезнью сердца (ИБС), перенёсших инсульт или инфаркт миокарда, вызывают дисфункцию тромбоцитов и повышают риск развития геморрагических осложне-ний. Практически всегда в этих случаях помогает назначение десмопрессина, за исключением ситуации применения ингибиторов СРНЬ/Ша. В последнем случае может возникнуть необходимость в трансфузии тромбоцитов.
Болезнь Виллебранда - наследуемый по аутосомно-доминантному типу гемор-рагический диатез, вызванный дефектом или отсутствием фактора Виллебранда в плазме. Фактор Виллебранда имеет две основные функции. Он необходим для образования тромбоцитарной пробки и защищает фактор VIII от расщепления в плазме. Существует три основных типа болезни Виллебранда (табл. 2-15).
Таблица 2-15. Болезнь Виллебранда: типы и терапия Фактор Виллебранда Терапия Количественный дефект Десмопрессин в большинстве случаев Качественный дефект Десмопрессин в легких случаях; концентраты фактора VIII, содержащие фактор Виллебранда; криопреципитат Полное отсутствие Концентраты фактора VIII, содержащие фактор Виллебранда, криопреципитат
Эпидемиологические исследования показали, что частота встречаемости болез-ни Виллебранда не превышает 1% в популяции, но диагностируют её обычно реже. Заболевание преобладает среди женщин с меноррагиями. У всех пациентов, страдающих кровоточивостью, следует определять количество фактора Виллебранда в крови.
Первый тип заболевания наиболее распространён, он составляет 70-80% всех лиц, страдающих болезнью Виллебранда. При этой форме заболевания снижено количество нормально функционирующего фактора Виллебранда. Тенденция к кровоточивости обычно выражена умеренно, но бывают и тяжёлые проявления.
Второй тип характеризуется качественным дефектом фактора Виллебранда, обусловленным мутацией в гене этого фактора.
Третий тип характеризуется отсутствием фактора Виллебранда и снижением концентрации фактора VIII в плазме обычно Диагностика основана на лабораторных тестах, главным образом на определе-нии концентрации и функции фактора Виллебранда и активности фактора VIII.
Терапия направлена на нормализацию трёх параметров - содержания факторов Виллебранда и VIII, времени кровотечения. Этого достигают назначением десмопрессина, стимулирующего эндогенный гемостаз, и введением концентрата фактора VIII, содержащего фактор Виллебранда. Десмопрессин эффективен при первом, реже при втором типе болезни Виллебранда и неэффективен при третьем типе. При внутривенном введении эффект практически молниеносный, при необходимости продолжения лечения повторные дозы вводят через 8-12 ч. При втором и третьем типах болезни Виллебранда и в случае отсутствия эффекта от назначения десмопрессина при первом типе терапию проводят концентратами фактора VIII, содержащими фактор Виллебранда. Чистые концентраты фактора VIII не используют, так как фактор VIII имеет очень короткий полупериод жизни в отсутствие фактора Виллебранда. Все концентраты фактора VIII, содержащие фактор Виллебранда, получают из плазмы, поэтому во избежание трансфузионного заражения вирусными инфекциями необходимо применять концентраты, прошедшие вирусную инактивацию. Именно вирусная небезопасность криопреципитата ограничивает его применение в терапии болезни Виллебранда. Иногда, особенно при выражен-ной кровоточивости слизистых оболочек, прибегают к назначению ингибитора фибринолиза - введению транексамовой кислоты.
Лицам с болезнью Виллебранда противопоказаны антикоагулянты, дезагреган- ты, переливание декстранов. Не следует назначать им внутримышечные инъекции, так как высок риск развития внутримышечных гематом.
Нарушения коагуляционного каскада
При заболеваниях, обусловленных патологией коагуляционного каскада, в конечном счёте нарушается образование фибрина. При врождённом характере заболевания отсутствует какой-либо один фактор свёртывания в плазме, в то время как при приобретённых заболеваниях наблюдают дефицит нескольких факторов (например, при вторичных нарушениях гемостаза вследствие болезней печени, дефицита витамина К, использовании антикоагулянтов).
ГЕМОФИЛИЯ АИВ
Гемофилия - врождённое заболевание системы свёртывания, наследуемое по рецессивному сцепленному с полом типу и обусловленное дефицитом фактора VIII (гемофилия А) или фактора IX (гемофилия В). Клинически две эти формы заболевания идентичны, их дифференцируют только при исследовании концентрации каждого фактора в крови.
Частота гемофилии А составляет один на 5000 новорождённых мальчиков, гемофилии В - один на 30 ООО новорождённых мальчиков. Соответственно 80% больных гемофилией имеют гемофилию А, 20% - гемофилию В.
Гены факторов VIII и IX сцеплены с женской Х-хромосомой, поэтому гемофилия поражает только мужчин, а женщины становятся носителями. У женщин- носителей риск иметь мальчика, больного гемофилией, составляет 25%, девочку- носителя - тоже 25%. У мужчин с гемофилией дочь всегда будет носителем, а сын всегда будет здоров.
Клинические проявления гемофилии зависят от концентрации фактора VIII или IX в крови. Их активность определяют в международных единицах (МЕ). В норме активность факторов VIII или IX составляет около 1 МЕ в 1 мл плазмы. Различают три степени тяжести гемофилии (табл. 2-16).
Таблица 2-16. Степень тяжести гемофилии Степень тяжести Концентрация фактора VIII, т/т Концентрация фактора IX, МЕ/дл Лёгкая гемофилия А 5-25 100 Умеренная гемофилия А 1-4 100 Тяжёлая гемофилия А В интенсивной терапии, как правило, нуждаются больные с тяжёлыми формами гемофилии, при которых спонтанные кровотечения и кровоизлияния могут возни-кать в любых органах и тканях, носить рецидивирующий характер. Кровоизлияния в суставы ведут к развитию гемофилической артропатии, в мышцы - могут вызвать синдром сдавления, в мягкие ткани - образование псевдоопухоли, которая инкапсулируется и, медленно увеличиваясь в результате повторных кровоизлияний, сдавливает окружающие ткани. Внутричерепное кровоизлияние - одна из самых частых причин смерти больных гемофилией.
Лёгкую форму гемофилии часто не распознают до зрелого возраста мальчиков, когда она может быть впервые обнаружена при необходимости проведения какого- либо оперативного вмешательства. Тяжёлую гемофилию обычно диагностируют на 1-2-м году жизни мальчиков, когда они начинают ходить. Первыми признаками могут быть кровоизлияния в суставы, подкожные гематомы вследствие детских шалостей или внутримышечных инъекций. В коагулограмме отмечают удлинение АЧТВ при нормальных показателях протромбинового и тромбинового времени, а также времени кровотечения. Диагноз подтверждают исследованием концентрации факторов VIII, IX и Виллебранда.
Для лечения используют концентраты факторов VIII или IX, получаемые из донорской плазмы или с помощью генной технологии. Современные концентраты факторов свёртывания вирусбезопасны и высокоэффективны. В расчёте дозы концентрата фактора исходят из необходимости поддержания гемостатической концентрации в крови реципиента не менее 50% в день операции и в течение
14 дней послеоперационного периода. Обычно вычисляют необходимое для разового введения количество МЕ фактора VIII или IX следующим образом:
объём крови (мл) = 7% массы тела (кг); например, 7% от 70 кг = 4900 мл;
объём плазмы (мл) = 60% объёма крови (мл); 60% от 4900 мл = 2940 мл;
количество МЕ фактора VIII = 50% объёма плазмы (мл); т.е. в нашем примере 1470 МЕ.
Кратность внутривенного введения для концентратов фактора VIII - каждые
ч, для фактора IX - каждые 12 ч.
Примерно у 30% больных гемофилией А в процессе лечения концентратами фактора VIII в крови появляются антитела, блокирующие прокоагулянтную активность препарата и поэтому названные ингибиторами. При гемофилии В частота появления ингибитора меньше. Титр ингибитора измеряют в единицах Бетесда (БЕ). Одна БЕ равна количеству ингибитора, ингибирующего 50% одной единицы фактора VIII, введённого в тест-систему. Низкий титр БЕ (5-10 БЕ) может быть преодолён увеличением дозы фактора, однако высокий титр БЕ (более 10 БЕ) делает невозможным получение гемостатического эффекта, так как всё количество введённого препарата будет блокировано антителами. Для удаления антител рекомендуют проведение плазмафереза (не следует замещать удаляемую плазму донорской СЗП, так как в ней находится фактор VIII), назначение глюкокорти- коидов. В последнее время широко стали использовать в подобных ситуациях рекомбинантный активированный фактор VII и активированный концентрат протромбинового комплекса. При проведении терапии таких больных обязательна консультация коагулолога.
ДЕФИЦИТ ФАКТОРА VII
Фактор свёртывания VII, связываясь с тканевым фактором, активируется и становится тем пусковым рычагом, с которого начинается процесс коагуляции. Генетический вариант врождённого дефицита фактора VII диагностируют очень редко. Клинически он проявляется образованием синяков при незначительных травмах, длительными некупируемыми носовыми и маточными кровотечениями. При тяжёлом дефиците фактора VII (менее 1%) могут возникать кровотечения, как при гемофилии, - гемартрозы, забрюшинные гематомы и внутримозговые кровоизлияния. При этом АЧТВ остаётся в пределах нормы, но протромбиновое время и МНО увеличиваются. Для окончательной диагностики необходимо исследование активности фактора VII в крови.
Приобретённый дефицит фактора VII развивается при заболеваниях печени, лечении варфарином, недостатке витамина К. При этом будут снижены в крови концентрации других зависимых от витамина К факторов свёртывания - II, IX, X.
Терапия носит заместительный характер. В качестве источника фактора VII используют переливание СЗП, назначают концентрат протромбинового комплекса. В последнее время в арсенале врача появился рекомбинантный активированный фактор VII, хорошо зарекомендовавший себя как в терапии дефицита фактора VII, так и в лечении ингибиторных форм гемофилии и тромбоцитопенических кровотечений. Для достижения гемостаза необходимо повысить концентрацию фактора VII в крови пациента до 15-20%. Рекомендуемая доза рекомбинантного активированного фактора VII составляет 90-120 мг/кг внутривенно через каждые
3 ч до остановки кровотечения. Необходим лабораторный мониторинг для контроля эффективности и уточнения дозы, а также для исключения возможного риска тромбоэмболических осложнений.
ДРУГИЕ ВРОЖДЁННЫЕ ДЕФИЦИТЫ ПЛАЗМЕННЫХ ФАКТОРОВ СВЁРТЫВАНИЯ
Дефицит фактора XI (гемофилия С) - врождённый геморрагический диатез, носящий выраженный этнический характер и диагностируемый преимущественно среди евреев и армян. Повторные кровотечения различной локализации (носовые, меноррагии, послеродовые, посттравматические) развиваются при содержании фактора XI менее 10%. АЧТВ удлинено, протромбиновое время в пределах нормы. При невозможности лабораторно верифицировать дефицит фактора XI при повышенной кровоточивости следует назначать переливание донорской СЗП. Назначение антифибринолитиков (транексамовой и аминокапроновой кислоты) малоэффективно, а при гематурии противопоказано.
Афибриногенемия - очень редкий геморрагический диатез, проявляющийся частыми спонтанными тяжёлыми кровотечениями. При гипофибриногенемии спонтанные кровотечения появляются только при концентрации фибриногена менее 0,5 г/л. АЧТВ и протромбиновое время значительно удлинены. Диагноз подтверждают лабораторным исследованием содержания фибриногена. Терапия - переливание СЗП, криопреципитата или концентрата фибриногена.
Дисфибриногенемии - гетерогенная группа наследственных заболеваний с дисфункцией фибриногена. Иногда эта дисфункция проявляется резистентностью фибринового сгустка к фибринолизу, чаще - повышенной чувствительностью к фибринолизу. В первом случае существует тенденция к повышенному тромбооб- разованию, во втором - к кровоточивости. Лечение после верификации диагноза (с привлечением коагулолога) - переливание СЗП, реже - назначение фибрино-гена.
Изолированный врождённый дефицит протромбина, факторов V, X,
XII, XIII в практике врача интенсивной терапии встречается крайне редко. Специфической клинической картины эти нарушения не имеют, для их диагно-стики необходимы не только скрининговые исследования коагулограммы, но и специфические анализы концентрации конкретных факторов свёртывания. При обнаружении этиологической причины кровотечения переливают СЗП.
АНТИФОСФОЛИПИДНЫЙ СИНДРОМ И ВОЛЧАНОЧНЫЙ АНТИКОАГУЛЯНТ
Кровоточивость слизистых оболочек в сочетании с проявлениями венозного или артериального тромбоза на фоне тромбоцитопении с одновременным обнару-жением в крови антифосфолипидных антител и/или волчаночного антикоагулянта характеризует антифосфолипидный синдром и его тяжёлую форму - катастрофический антифосфолипидный синдром. Данный синдром возникает при системной красной волчанке, коллагенозах, онкологических заболеваниях, инфекциях и беременности. Многие случаи катастрофического антифосфолипидного синдрома носят идиопатический характер. В основе этого синдрома лежит образование аутоантител, направленных против фосфолипидов, в частности кардиолипина. Последний образует комплекс с белком (32-гликопротеином-1 (р2-СР1), который входит в состав протромбина. По этой причине плазма больных с катастрофическим антифосфолипидным синдромом обладает антикоагулянтной активностью, и по содержанию «волчаночного антикоагулянта» можно судить о степени этой активности. При антифосфолипидном синдроме будут пролонгированы все зависимые от протромбина тесты. При этом, несмотря на удлинение АЧТВ т УИГО, время свёртывания т У1УО не нарушено, что и определяет высокий риск тромбозов при антифосфолипидном синдроме.
Терапия катастрофического антифосфолипидного синдрома носит комплексный характер. На фоне проведения плазмафереза назначают НФГ, преднизолон. При неэффективности проводят пульс-терапию метилпреднизолоном. Удаляемую плазму восполняют донорской СЗП. Нередко при тотальных тромбозах мезентериальных сосудов приходится прибегать к хирургическому вмешательству, существенно ухудшающему прогноз.
ЦИРРОЗ ПЕЧЕНИ
Комплексное нарушение гемостатического баланса вследствие снижения синтеза многих факторов свёртывания и ингибиторов при заболеваниях печени сопровождается развитием патологической кровоточивости. При этом снижается количество и функция тромбоцитов, увеличивается фибринолиз, удлиняется протромбиновое время, повышается МНО. Часто спленомегалия, наблюдаемая при циррозе печени, усугубляет тромбоцитопению. В случае тяжёлого кровотечения или хирургического вмешательства назначают переливание СЗП, введение рекомбинантного активированного фактора VII, протромбинового комплекса. Транексамовая кислота и десмопрессин действуют кратковременно.
Диссеминированное внутрисосудистое свёртывание
ДВС - полное нарушение взаимодействия всех систем гемостатического баланса, определяющих его равновесие, к которым относят эндотелиальные клетки, тромбоцитарный (первичный) гемостаз, плазменную систему свёртывания и фибринолиз. Для ДВС характерно одновременное наличие кровоточивости и микротромбирования, приводящих к быстрому развитию органной и ПОН. Расшифровка феномена ДВС (в которой в нашей стране ведущую роль сыграли работы М.С. Мачабели и З.С. Баркагана) объяснила многие казавшиеся раз-розненными явления, сопровождающие такие формально разные критические состояния, как сепсис, массивная кровопотеря, ожоги, укусы змей, осложнения беременности и др.
В практике интенсивной терапии ДВС чаще всего развивается на фоне сепсиса, массивной кровопотери (особенно в акушерстве), травм, онкологических и гематологических заболеваний.
ЭТИОЛОГИЯ И МЕХАНИЗМ РАЗВИТИЯ
Развитие ДВС может быть обусловлено различными причинами и состояниями, как это представлено ниже. Чаще ДВС принимает характер острого (молниеносного) течения, реже наблюдают хроническое течение.
Причины развития ДВС-синдрома:
Шок (гиповолемия и гипоксия) любой этиологии.
Инфекции.
Сепсис.
Бактериальный.
Вирусный.
Грибковый.
Травма
Ожоги.
Краш-синдром.
-ЧМТ.
Жировая эмболия.
Большие хирургические операции.
Осложнения беременности и родов
Тяжёлая эклампсия.
Отслойка плаценты.
Внутриутробная смерть плода.
Эмболия околоплодными водами.
НЕНР-синдром.
Анафилаксия
Инсульт
Острый внутрисосудистый гемолиз
Протезирование сосудов
Укусы змей
Новообразования
Аденокарцинома.
Гемобластозы.
Болезни печени
Цирроз.
Острый фульминантный гепатит.
Патологическая активация звеньев системы гемостаза может быть вызвана различными факторами. Активация эндотелиальных клеток эндотоксином при инфекциях, отравлениях, ацидозе и гипоксемии сопровождается появлением на поверхности их мембраны тканевого фактора. Плазменная система свёртывания активируется высвобождением тканевого фактора в результате травмы, сепсиса, осложнений беременности, бластемии при гемобластозах.
Системная активация свёртывания приводит к образованию в русле крови чрезмерного количества циркулирующего тромбина, который вызывает генерализованное образование фибрина, активацию и потребление факторов VIII и V, а также активацию тромбоцитов. В результате запускается процесс массированного микротромбообразования в системе микроциркуляции, ведущий к развитию ПОН и усугубляющий повреждение эндотелиальных клеток. С другой стороны, гене-рализованное фибринообразование активирует фибринолитическую систему, из активированных эндотелиальных клеток, тромбоцитов и лейкоцитов выделяются активаторы фибринолиза, которые активно потребляются.
Такая массивная системная активация гемостаза приводит к быстрому потреблению всех факторов свёртывания, тромбоцитов и ингибиторов. Потребление тромбоцитов при ДВС может идти экстремально быстро, костномозговая их продукция не успевает пополнять циркулирующий пул тромбоцитов. К тому же циркулирующие тромбоциты становятся функционально дефектными в результате воздействия продуктов деградации фибрина. При болезнях печени сниженный синтез белков вследствие печёночной недостаточности способствует углублению дефицита факторов свёртывания и их ингибиторов. Кроме того, имеет значение патологическое разрушение факторов свёртывания бактериальными протеазами при сепсисе, энзимами поджелудочной железы при панкреонекрозе или около-плодной жидкости при эмболии околоплодными водами. Результатом этих про-цессов становится кровоточивость, обусловленная тромбоцитопенией, гиперфи- бринолизом и дефицитом факторов свёртывания.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Спектр клинических проявлений ДВС зависит как от причины, его вызвавшей (ДВС всегда вторично, это не нозологическая форма, а синдром, всегда ассоциированный с каким-либо основным заболеванием), так и от условий, сопутствующих его развитию. При остром течении ДВС-синдрома его проявлениями могут быть признаки ПОН, указывающие на поражение ЦНС, почечную, печёночную, лёгочную дисфункции. Характерны метаболический ацидоз, протеинурия, гипоксия, гипотензия и лихорадка. Геморрагические признаки включают петехии и экхи- мозы на коже, спонтанную кровоточивость слизистых оболочек, кровотечение из мест инъекций и хирургических ран, в тяжёлых случаях - внутримозговые кровоизлияния. Микротромбозы вызывают ишемическое повреждение внутренних органов, главным образом головного мозга, лёгких, почек и печени. Тромбоз сосудов дермы и субдермы сопровождается акроцианозом, возможным развитием гангрены пальцев рук или стоп. Ишемические признаки ДВС проявляются неврологическими нарушениями сознания (заторможенность, быстрая истощаемость и односложность в ответах на вопросы), гипоксемией и гипоксией с нарушением частоты и ритма дыхания, олигурией или анурией, гипоальбуминемией и гипопро- тромбинемией.
ДИАГНОСТИКА
Первичная диагностика ДВС-синдрома полностью основана на клинической картине, включающей геморрагические и ишемические признаки. Различают гипер- и гипокоагуляционные фазы острого ДВС-синдрома, при этом гиперкоагу- ляционную фазу ДВС-синдрома необходимо дифференцировать от хронического гиперкоагуляционного синдрома (статуса), который принципиально отличается от ДВС-синдрома как патогенетически, так и по клиническим и лабораторным данным.
Гиперкоагуляционный синдром - повышенная готовность крови к свёртыванию, компенсированная противосвёртывающими механизмами. При нём нет ни локальных, ни диссеминированных тромбов в сосудистой системе, соответственно нет клинических проявлений тромбозов. Но в лабораторных данных отмечают укорочение АЧТВ, протромбинового времени, повышенную активность тромбо-цитов, снижение фибринолиза, быстрое образование сгустка в пробирке.
Гиперкоагуляционная фаза ДВС-синдрома часто скоротечна, и врач её может не диагностировать. Появляются клинические признаки органной ишемии. Все лабораторные признаки гиперкоагуляции (АЧТВ, протромбин, активация тромбо-цитов) выражены, но одновременно с этим появляются первые начальные призна-ки потребления факторов свёртывания - нерезко снижается количество тромбо-цитов, концентрация антитромбина III, протеина С. Сгусток в пробирке образуется достаточно быстро, но он рыхлый и нестойкий. Важный симптом - часто быстрое тромбирование иглы или катетера при внутривенной пункции.
Гипокоагуляционная фаза ДВС-синдрома характеризуется признаками диффуз-ного геморрагического диатеза (кровоточивость петехиально-экхимозного типа) и лабораторными маркёрами потребления факторов свёртывания системы гемо-стаза - удлинением времени кровотечения, АЧТВ, протромбинового времени, существенным снижением количества тромбоцитов и их дисфункцией, уменьше-нием концентрации фибриногена, фактора VIII, появлением Б-димеров.
ЛЕЧЕНИЕ
В основе терапии ДВС лежит купирование запустившего его пускового патологического процесса. Однако этиологическая терапия этих заболеваний (антибиотики при сепсисе, химиотерапия или операция при опухолях, плазмаферез при остром внутрисосудистом гемолизе и др.) даёт эффект лишь через определённый промежуток времени. Вот почему столь важны сопутствующие терапевтические мероприятия, обеспечивающие восстановление и поддержание ОЦК, адекватную оксигенацию, коррекцию гипотонии с целью улучшения микроциркуляции.
В то же время критическое состояние больного, усугубляемое проявлениями ДВС, делает необходимым быструю трансфузионную коррекцию нарушений в системе гемостаза.
В терапии гиперкоагуляционной фазы ДВС-синдрома, когда есть клинические проявления ишемии органов вследствие микротромбирования, назначают гепарин натрия. Гепарин натрия ингибирует активность тромбина, уменьшая тем самым образование фибрина. Обычно назначают 8-10 МЕДкгхч) при постоянном вну-тривенном введении с помощью дозатора лекарственных веществ (инфузомата). Следует помнить, что гепарин натрия эффективно работает при условии достаточного количества в плазме крови антитромбина III. При его снижении необходимо переливание СЗП (10 мл/кг) или назначение коммерческих препаратов антитромбина III (до 3000 МЕ/сут). Критерием эффективности терапии гепарином натрия будет снижение концентрации продуктов деградации фибрина и Б-димеров, повышение содержания фибриногена, укорочение протромбинового времени. Использование в этой ситуации низкомолекулярных гепаринов нецелесообразно из-за их недостаточной эффективности и невозможности мониторинга. В терапии ДВС-синдрома септической этиологии хорошо себя зарекомендовал активированный протеин С, который, будучи антикоагулянтом, ингибирует активацию факторов V и VIII и уменьшает образование тромбина.
Значительно чаще врач интенсивной терапии встречается с гипокоагуляционной фазой ДВС-синдрома, в трансфузионной терапии которой ведущая роль принадлежит восполнению дефицита факторов свёртывания. ДВС - комплексное нарушение системы гемостаза, поэтому «первую скрипку» в его лечении играет переливание СЗП - комплексной трансфузионной среды, в которой в оптимальном наборе есть все необходимые факторы свёртывания. Цель переливания СЗП - повысить концентрацию фибриногена выше 1-1,5 г/л. Терапевтической дозой считают переливание СЗП из расчёта 15-20 мл/кг. При отсутствии достижения гемостаза возможно повторное введение СЗП под лабораторным контролем за концентрацией факторов свёртывания. Иногда необходимо добавление к переливанию СЗП препаратов антитромбина III, особенно если его концентрация в крови менее 70%. При угрозе циркуляторной перегрузки с целью уменьшения объёма прибегают к переливанию криопреципитата (одна доза на 10 кг массы тела). Применение плазмафереза в терапии ДВС-синдрома направлено как на терапию основного заболевания (например, острого внутрисосудистого гемолиза вследствие переливания несовместимых по антигенам системы АВО эритроцитов), так и на предупреждение циркуляторной перегрузки при необходимости переливания больших объёмов плазмы.
Переливание тромбоцитов при ДВС-синдроме показано только при развитии кровотечения и снижении их количества менее 50х109/л. Цель переливания тромбоцитов - превысить это значение, для чего необходима, как правило, одна единица концентрата тромбоцитов (55-70х109) на 10 кг массы тела за одну трансфузию. При выраженном потреблении тромбоцитов необходимы повторные переливания каждые 24 ч.
Переливание эритроцитов показано только по жизненным показаниям при верифицированных признаках гипоксемии и тканевой гипоксии вследствие нехватки эритроцитов в русле крови.
Важно помнить, что некоторые пациенты с лабораторными признаками ДВС не имеют клинических проявлений тромбоза или тенденции к кровоточивости. Такие пациенты не нуждаются в трансфузионной коррекции гемостаза, им необходимо проводить терапию по основному заболеванию.
СПИСОК ЛИТЕРАТУРЫ ^
Практическая трансфузиология / Под ред. Г.И. Козинца. - М.: Практическая медицина, 2005. - 544 с.
Шевченко Ю.Л., Шабалин В.Н., Заривчацкий М.Ф., Селиванов Е.А. Руководство по общей и клинической трансфузиологии. - СПб.: Фолиант, 2003. - 608 с.
КН^аагс! Т., ТаЬапега У.Р.К., Войжс! К. е* а1. РНагтасокте1лс$ о^ гесотЫпаШ: асПуа^ес! ^асШг VII т 1гаита райеп^ т1\\ зеуеге Ыеес11п§ // Сп1. Саге. - 2006. - Уо1. 10. - Р. 104.
МагИпошИг II., Кепе* С., 5е§а1 Е. е1 а1. КесошЫпап!; асПуа^ес! Гас^ог VII Ьг афипсйуе Ьетог- гНа§е соп1го1т 1;гаита // ]. Тгаита. - 2001. - Уо1. 51. - Р. 431-438.
Утсеп!: ].Ь., Ко$$ат1: К., Кюи В. е* а1. Кесоттепс1а1:юп$ оп 1Ье и$е о!" гесотЬтап! асй- уа*ес1 ^ас1;ог VII аз ап аф"ипсНуе 1;геа1:теп1; Гог та$$1уе Ыеес11п§ - а Еигореап регзресИуе // Сп{. Саге. - 2006. - Уо1. 10. - Р. 120.
К данной группе относят генетически детерминированные гипокоагуляции, характеризующиеся дефицитом, а также молекулярными аномалиями факторов свертывания крови.
Так, 83-90% всех наследственных нарушений свертываемости крови представляют 2 разновидности дефицита фактора VTII гемофилию А (70-78%) и болезнь Виллебранда (9-18%); еще 6-13% связаны с дефицитом фактора IX (гемофилия В). Таким образом, на дефицит только двух факторов свертывания - VIII и IX - приходится около 96-98% всех наследственных коагулопатий. Дефицит факторов VII и V регистрируется в 0,5-1,5%, фактора X - в 0,3-0,5% случаев.
Не все нарушения в свертывающей системе крови сопровождаются кровоточивостью: она может отсутствовать или быть слабо выраженной.
Гемофилия А . Данное заболевание представляет собой наиболее распространенную коагулопатию, в основе которой лежит дефицит фактора VIII (антигемофильного глобулина), и является единственной среди них формой с рецессивным, сцепленным с X-хромосомой наследованием.
Разнообразие форм патологии фактора VIII отражает сложность его структуры. В крови фактор VIII циркулирует в виде протеинового комплекса, состоящего из ряда однотипных субъединиц.
Наследование . Ген гемофилии, располагающийся в Х-хромосоме, от больного мужчины наследуется всеми его дочерьми, которые в дальнейшем неизбежно являются носителями заболевания, при этом сыновья больного остаются здоровыми (в связи с тем, что получают Х-хромосому от здоровой матери).
Также следует отметить, что у женщины - носителя гемофилии имеется возможность в 50% случаев родить здорового сына, а половина дочерей становятся носителями гена гемофилии.
Женщины-носители, как правило, кровоточивостью не страдают, поскольку вторая нормальная Х-хромосома обеспечивает синтез фактора VIII, чего в большинстве случаев достаточно для обеспечения гемостаза.
Однако норма фактора VIII варьирует в очень больших пределах (60-250%). В связи с этим у некоторых передатчиц уровень фактора VIII в плазме может составлять 11-20%, что создает угрозу кровотечений при травмах, операциях и в родах. Об этой опасности врачу следует помнить при хирургических вмешательствах у матерей, сестер и особенно дочерей больных гемофилией. Перед операцией и перед родами у них следует проверять уровень фактора VIII в плазме и при показателях ниже 25% профилактически вводить криопреципитат по 7-10 ЕД/кг в сутки.
Выявлению носительства гена гемофилии способствует детальное изучение семейного геморрагического анамнеза у всех кровных родственников больного по материнской линии.
Наследственный генез устанавливается при гемофилии А в 70-75% случаев, а при гемофилии В - в 90-91%. Ген гемофилии А, несомненно, часто мутирует, поскольку число больных за много веков не уменьшилось, хотя до недавнего времени значительная часть их умирала до достижения детородного возраста, что вело к естественной убыли аномальных Х-хромосом.
Симптомы Нарушений коагуляционного гемостаза
Степень выраженности кровотечений зависит от дефицита фактора VIII в плазме, содержание которого в различных гемофилических семьях генетически запрограммировано.
Уровень факторов, обладающих четким антигемофильным действием (VIII или IX), составляющий от 0 до 1%, обусловливает крайне тяжелое течение рассматриваемой патологии, уровень факторов от 1 до 2% обусловливает тяжелое, от 2 до 5% -среднетяжелое, а свыше 5% - легкое течение заболевания. В последнем случае существует вероятность развития кровотечений, представляющих серьезную опасность для жизни больного, что имеет особое значение при проведении у него различных оперативных вмешательств либо в случае травмирования.
В клинической картине гемофилии преобладают кровоизлияния в крупные суставы конечностей, глубокие подкожные, межмышечные и внутримышечные гематомы, обильные и длительные кровотечения при травмах, гематурия (появление крови в моче). Реже наблюдаются другие кровотечения, в том числе и такие тяжелые и опасные, как забрюшинные гематомы, кровоизлияния в органы брюшной полости, желудочно-кишечные кровотечения внутричерепные кровоизлияния.
Прослеживается отчетливая возрастная эволюция проявлений болезни. При рождении могут наблюдаться более или менее обширные кефалогематомы (кровоизлияние под надкостницу костей черепа), подкожные и внутрикожные кровоизлияния, поздние кровотечения из пупочного канатика. Иногда болезнь выявляется при первой внутримышечной инъекции, которая может стать причиной большой, опасной для жизни межмышечной гематомы. Прорезывание зубов часто сопровождается не очень обильными кровотечениями.
В первые годы жизни часто бывают кровотечения из слизистой оболочки полости рта, связанные с травмой различными острыми предметами. Когда ребенок учится ходить, падения и ушибы часто сопровождаются обильными носовыми кровотечениями и гематомами на голове, кровоизлияниями в глазницу, а также заглазничными гематомами, которые могут привести к потере зрения. У ребенка начавшего ползать, типичны кровоизлияния в области ягодиц.
Затем на первый план выступают кровоизлияния в крупные суставы конечностей. Острые кровоизлияния в суставы появляются тем раньше, чем тяжелее гемофилия. Первые кровоизлияния предрасполагают к повторным в те же суставы. У каждого больного с особым упорством и частотой поражаются кровоизлияниями I - III сустава. Это связано с морфологической перестройкой и вторичными воспалительными изменениями тканей сустава.
Установлено, что синовиальная оболочка является главным, а возможно, и единственным источником кровоизлияния в сустав, поскольку после полной синовэктомии (удаления синовиальной оболочки) такие кровоизлияния п-екращаются и не повторяются. Наиболее часто поражаются коленные суставы, за ними голеностопные и локтевые, а затем со значительным перепадом - тазобедренные. Сравнительно редко наблюдаются кровоизлияния в мелкие суставы кистей и стоп (менее 1% всех поражений) и межпозвонковые суставы. У каждого больного в зависимости от возраста и тяжести заболевания поражаются от I - II до VIII - XII суставов.
Клинически важно различать острые кровоизлияния в суставы (первичные и повторяющиеся), хронические геморрагически-деструктивные остеоартрозы (артропатии), вторичный иммунный ревматоидный синдром как осложнение основного процесса.
Острый гемартроз - внезапное появление (часто после небольшой травмы) или резкое усиление боли в суставе. Кожа в области сустава красная и горячая на ощупь. Боль быстро (в течение нескольких часов) ослабляется после первого же переливания криопреципитата или антигемофильной плазмы и почти немедленно проходит при одновременном удалении крови из сустава. Если болевой синдром при таком лечении не ликвидируется, то следует искать дополнительную патологию - внутрисуставной перелом, отрыв мыщелка, ущемление ткани.
Остеоартрозы разделяются по стадиям на основе клинико-рентгенологических данных. В классификации выделяются 4 стадии поражения суставов.
В I, или ранней, стадии может быть увеличен объем сустава (с расширением суставной щели) в результате кровоизлияния. В «холодном» периоде функция сустава не нарушена, но рентгенологически может обнаруживаться утолщение и уплотнение суставной капсулы вследствие инфильтрации ее гемосидерином.
Во II стадии выявляются типичные изменения в подхрящевом отделе эпифизов -краевые узоры, образование одиночных овальных мелких ячеистых деструкции и кист. Более выражен остеопороз, суставная щель сохранена, но может быть умеренно суженной.
В III стадии сустав резко увеличен, деформирован, имеет неровную и бугристую структуру, определяется выраженная гипотрофия мускулатуры. Подвижность пораженных суставов более или менее ограничена, что связано как с поражением самого сустава, так и с изменениями мышц и сухожилий. Рентгенологически суставы утолщены, резко деформированы, суставные поверхности уплощены, эпифизы расширены за счет разрастания костной ткани, диафизы уменьшены, суставная щель сужена. Выражен остеопороз, легко возникают внутрисуставные переломы. В бедренной кости отмечается типичное для гемофилии кратеро- или туннелеподобное разрушение костного вещества в области межмыщелковой ямки. Надколенник частично разрушается. Внутрисуставные хрящи разрушены, в полости сустава обнаруживаются подвижные, нередко замурованные в старые организовавшиеся сгустки крови осколки этих хрящей. Возможны различного рода подвывихи и смещения костей.
В IV стадии функция сустава почти полностью утрачивается, суставная щель сужена, плохо визуализируется на рентгенограмме и часто заращена соединительной тканью. Выражен склероз околохрящевых отделов кости, сочетающийся с образованием щелей и формированием кист в области эпифизов. Возможны патологические внутрисуставные переломы. Костные анкилозы исключительно редки и, по сути дела, никогда не наблюдаются, если только в прошлом не проводилось неправильное лечение (с длительной иммобилизацией конечности).
С возрастом тяжесть и распространенность суставного поражения неуклонно прогрессируют и усугубляются возникновением околосуставных гематом.
Вторичный ревматоидный синдром (синдром Баркагана - Егоровой), впервые описанный авторами в 1969 г., является частой формой поражения суставов у больных гемофилией. Во многих случаях он просматривается, поскольку наслаивается на предшествующие кровоизлияния в суставы и свойственные гемофилии деструктивные процессы в суставах. Внимательное обследование больных позволяет легко диагностировать вторичный ревматоидный синдром, что имеет существенное значение для дальнейшего правильного лечения. Этот синдром сопровождается хроническим воспалительным процессом (часто симметричным) в мелких суставах кистей и стоп, не поражавшихся ранее кровоизлияниями, с последующей их типичной деформацией, болью в крупных суставах, не купирующейся, а нередко и обостряющейся после переливаний плазмы и введений криопреципитата. Также данный синдром протекает с выраженной утренней скованностью в суставах, неуклонным прогрессированием суставного процесса вне связи со свежими кровоизлияниями, появлением или резким усилением лабораторных признаков воспалительного процесса, в том числе и иммунологических, - с повышением уровня глобулинов в сыворотке крови, сиаловых кислот, фибриногена, нарастанием концентрации циркулирующих иммунных комплексов и в ряде случаев - титра ревматоидного фактора. У большинства больных синдром появляется в возрасте старше 10-14 лет, к 20 годам его частота доходит до 5,9%, а к 30 - до 13%.
С возрастом распространенность и тяжесть всех поражений суставов неуклонно прогрессируют, что приводит к инвалидности, заставляет больных пользоваться костылями, колясками и другими приспособлениями.
Прогрессирование поражения суставов зависит от частоты острых кровоизлияний в суставы, своевременности и полноценности их лечения (очень важно раннее переливание кровезаменителей), качества ортопедической помощи больному, правильного применения лечебной физкультуры, физиотерапевтических и бальнеологических воздействий, выбора профессии и ряда других обстоятельств. В настоящее время все эти вопросы чрезвычайно актуальны, поскольку продолжительность жизни больных гемофилией благодаря успехам коррекционной терапии резко возросла.
Довольно тяжело протекают и представляют опасность для больного следующие виды обширных и напряженных гематом: подкожные, межмышечные, субфасциальные и забрюшинные. Постепенно увеличиваясь, они могут достигать огромных размеров, содержать 0,5-3 л крови и более, приводить к развитию анемии у больных, вызывать компрессию (сдавление) и деструкцию (разрушение) окружающих тканей и питающих их сосудов, некроз. Так, например, забрюшинные гематомы нередко полностью разрушают большие участки тазовых костей (диаметр зоны деструкции - до 15 см и более), гематомы на ногах и руках разрушают трубчатые кости, пяточную кость. Гибель костной ткани обусловливают также кровоизлияния под надкостницу. Эти деструкции костей на рентгенограммах имеют сходство с опухолевыми разрушениями (например, при остеосаркомах). Нередко гематомы кальцинируются, а иногда приводят к образованию новых костей (остеонеогенез). Они могут замыкать суставы и полностью их обездвиживать.
Многие гематомы, оказывая давление на нервные стволы или мышцы, вызывают параличи, нарушения подвижности, чувствительности, быстро прогрессирующую атрофию мышц. Для кровоизлияний в область подвздошно-поясничной мышцы особенно характерны сгибательные движения бедра. Особое внимание уделяется тем гематомам, которые способны вызывать развитие стеноза верхних дыхательных путей. К таким гематомам относятся гематомы мягких тканей подчелюстной области, гематомы области шеи, зева и глотки.
У 14-30% всех больных с гемофилией развиваются обильные и длительные почечные кровотечения, которые создают серьезную угрозу жизни больного и с трудом поддаются лечению. Такие кровотечения могут возникать как спонтанно, так и в связи с травмами поясничной области, сопутствующими пиелонефритами, и, возможно, вследствие повышенного выделения кальция с мочой из-за деструкции костной ткани у больных гемофилией. Появлению или усилению таких кровотечений могут способствовать прием анальгетиков (ацетилсалициловая кислота и др.), обильные переливания крови и плазмы, приводящие к развитию вторичной тромбоцитопатии за счет дополнительного отрицательного воздействия на почки. Почечным кровотечениям часто предшествует длительная микрогематурия (малое количество эритроцитов в моче), которая регистрируется и в промежутках между эпизодами макрогематурии (большое количество эритроцитов в моче, заметное на глаз).
Появление крови в моче часто сопровождается выраженными дизурическими явлениями, приступами почечной колики, обусловленными образованием сгустков крови в мочевыводящих путях. Особенно интенсивны и выражены эти явления при лечении больных, когда временно восстанавливается нормальный гемостаз. Прекращению гематурии часто предшествует почечная колика, а нередко - и временное отсутствие мочи с азотемией.
Почечные кровотечения склонны к возобновлению, что с годами может привести к тяжелым дистрофически-деструктивным изменениям в этом органе, вторичной инфекции и амилоидозу, смерти от уремии (попадания продуктов обмена веществ, в норме выводимых с мочой, в кровь).
Обильные кровотечения из желудочно-кишечного тракта у больных гемофилией могут возникать спонтанно, хотя в большинстве случаев они провоцируются приемом ацетилсалициловой кислоты, бутадиона и других препаратов. Вторым источником кровотечений служат клинически выраженные или «скрытые» язвы желудка или двенадцатиперстной кишки, а также эрозивные гастриты различного происхождения. Вместе с тем иногда наблюдаются диффузные капиллярные кровотечения без каких-либо деструктивных изменений слизистой оболочки. Эти диапедезные кровотечения, при которых стенка кишечника на большом протяжении пропитывается кровью, быстро приводят к анемической коме, острой сосудистой недостаточности и летальному исходу.
Кровоизлияния в брыжейку, а также в большой и малый сальник зачастую создают ложное впечатление о развитии у больного острой хирургической патологии органов брюшной полости, такой как острый аппендицит, кишечная непроходимость, что особенно выражено в случае кровоизлияния под серозную оболочку в стенке кишки. Единственным ориентиром в подобных ситуациях может быть быстрая эффективность интенсивной заместительной терапии. Мгновенное начало такой терапии рекомендуется в любом случае - как для устранения кровотечений, так и в порядке подготовки больного к операции. Далее все решает результат лечения. Если вслед за струйным введением концентрата фактора VIII (или IX) болевой синдром и другие признаки острого живота быстро стихают, то можно продолжить наблюдение за больным при продолжающейся интенсивной заместительной терапии (неосложненное внутреннее кровоизлияние). Если эффект заместительной терапии выражен недостаточно, то необходимо хирургическое вмешательство.
Кровоизлияния в головной и спинной мозг и их оболочки при гемофилии почти всегда связаны либо с травмами, либо с приемом препаратов, нарушающих гемостатическую функцию тромбоцитов. Между моментом травмы и развитием кровоизлияния может быть светлый промежуток продолжительностью от 1-2 ч до суток.
Характерным симптомом, отличающим гемофилию от другой патологии, является длительное кровотечение в случае травмы и операции. Рваные раны значительно опаснее линейных разрывов. Кровотечения часто возникают не сразу после травмы, а через 1-5 ч.
Тонзиллэктомия (удаление небных миндалин) при гемофилии значительно более опасна, чем полостные хирургические операции.
Удаление зубов, особенно коренных, часто сопровождается многодневными анемизирующими кровотечениями не только из зубных лунок, но и из гематом, образовавшихся на месте инфильтрации тканей новокаином. Эти гематомы вызывают деструкцию челюсти. При гемофилии зубы следует удалять на фоне действия антигемофилических препаратов под общим наркозом. Удаление нескольких зубов лучше проводить одномоментно.
Часть осложнений при гемофилии обусловлена потерей крови, сдавлением и деструкцией тканей гематомами, инфицированием гематом. Большая группа осложнений связана также с иммунными нарушениями. Наиболее опасным из них является обнаружение в крови больных большого количества антител против фактора VIII свертывания крови (или IX), модифицирующих гемофилию в так называемую ингибиторную форму, при которой основной метод лечения - трансфузионная терапия -почти полностью утрачивает свою эффективность. Более того, повторное введение антигемофилических препаратов часто вызывает у больных быстрое нарастание количества данных антител, вследствие чего трансфузионная терапия, первоначально дававшая какой-то эффект, вскоре становится бесполезной.
Частота ингибиторной формы гемофилии колеблется от 1 до 20%, чаще - от 5 до 15%. При тяжелых формах гемофилии ингибиторы появляются в крови больных несравненно чаще, чем при легких, а у лиц старше 12 лет - намного чаще, чем в более раннем возрасте. При ингибиторных формах заметно нарушается гемостатическая функция тромбоцитов, учащаются кровоизлияния в суставы, кровь в моче, достоверно выше поражение суставов.
Из других иммуноаллергических нарушений иногда наблюдаются тромбоцитопения, изредка сочетающаяся с лейкопенией, аутоиммунная гемолитическая анемия с положительной пробой Кумбса, большая эозинофилия, амилоидоз почек.
Диагностика Нарушений коагуляционного гемостаза
Гемофилия диагностируется у всех больных с гематомным типом кровоточивости и поражением опорно-двигательного аппарата, а также при упорных поздних кровотечениях при операциях. Для ориентировочной диагностики решающее значение имеет выявление снижения интенсивности коагуляции (свертывания) крови в таких общих пробах, как время свертывания крови, активированное парциальное тромбопластиновое время, и в аутокоагуляционном тесте при нормальных показателях тромбинового и протромбинового времени.
С целью определения того, какой из факторов свертывания крови находится в дефиците, прибегают к помощи коррекционных проб, используя тест генерации тромбопластина или аутокоагуляционный тест.
Вид гемофилии можно идентифицировать и «тестами смешивания»: к плазме обследуемого больного последовательно в разных пробирках добавляют образцы плазмы, в которых отсутствует один из факторов свертывания (VIII, IX или XI). Отсутствие нормализации свертывания в одной из пробирок указывает на дефицит того же фактора в обеих смешиваемых плазмах, т. е. на его дефицит у больного.
Диагностика гемофилии заканчивается определением дефицита фактора в количественном отношении, что имеет значение для правильной оценки тяжести заболевания и проведения заместительной терапии.
Лечение Нарушений коагуляционного гемостаза
Основным методом лечения и профилактики гемофилических кровотечений любой локализации и любого происхождения является внутривенное введение достаточных доз препаратов крови, содержащих фактор VIII. Фактор VIII изменчив и практически не сохраняется в консервированной крови, нативной и сухой плазме. Для заместительного лечения пригодны только прямые переливания крови от донора к больному гемофилией, а также внутривенные вливания препаратов крови с сохраненным фактором VIII (антигемофильная плазма, криопреципитат, концентраты фактора VIII разной очистки).
К прямым переливаниям от донора прибегают лишь тогда, когда врач не располагает какими-либо другими антигемофилическими препаратами. Грубой ошибкой является переливание крови от матери больного, так как она - носительница болезни и уровень фактора VIII у нее резко снижен.
Ввиду короткого периода полувыведения фактора VIII в крови больного (около 6-8 ч) переливания крови, как и переливания антигемофильной плазмы, должны повторяться не реже 3 раз в сутки. Для остановки массивных кровотечений и надежного прикрытия различных хирургических вмешательств, когда уровень антигемофильного фактора должен поддерживаться выше 30-40%, такие переливания крови и плазмы непригодны. Хотя время свертывания и время рекальцификации (насыщения крови кальцием) нормализуется у больных гемофилией при повышении концентрации фактора VIII до 3-4%, этого уровня недостаточно для предупреждения кровотечений при операциях. Следовательно, при лечении и предоперационной подготовке следует ориентироваться только на количественное определение фактора VIII (либо на аутокоагулограмму), но не на показатели общего времени свертывания, теста потребления протромбина и других методик с низким порогом чувствительности.
Равный объем антигемофильной плазмы приблизительно в 3-4 раза эффективнее свежей консервированной крови. В разовых дозах 10-15 мл/кг и в суточных 30-50 мл/кг, разделенных на 3 части (первая доза в 1,5 раза больше 2 последующих), антигемофильная плазма позволяет недолго поддерживать 10-15%-ный уровень фактора VIII. Главная опасность такого лечения - перегрузка кровообращения больного объемом, что может привести к развитию отека легкого. Использование антигемофильной плазмы в концентрированном виде не меняет ситуации, так как высокая концентрация вводимого альбумина (белка) вызывает интенсивное перемещение жидкости из тканей в кровь, вследствие чего объем циркулирующей крови увеличивается так же, как и при переливании плазмы в нормальном разведении. Концентрированная сухая антигемоVIIильная плазма имеет лишь то преимущество, что в ней более концентрирован фактор С и в малом объеме он быстрее вводится в кровоток больного. Сухую антигемофильную плазму перед употреблением разводят дистиллированной водой до 1/3-1/2 исходного объема. Лечения антигемофильной плазмой вполне достаточно для купирования большинства острых кровоизлияний в суставы (кроме наиболее тяжелых), профилактики и лечения небольших кровотечений.
Наиболее надежны и эффективны при гемофилии концентраты фактора VIII. Самым доступным из них остается криопреципитат - выделяемый из плазмы с помощью охлаждения (криоосаждения) белковый концентрат, в котором достаточно фактора VIII, фибриногена и фактора XIII, но мало альбумина и ряда других белков. Низкое содержание в препарате альбумина позволяет вводить его в кровоток больных в очень больших количествах и увеличивать концентрацию фактора VIII до 100% и более, не опасаясь перегрузки кровообращения и отека легких. Основной недостаток криопреципитата - его нестандартность по активности.
Криопреципитат нужно хранить при -20°С, что затрудняет его транспортировку. При оттаивании препарат быстро утрачивает активность. Этих недостатков лишены сухой криопреципитат и современные концентраты фактора VIII. Их можно хранить в обычном холодильнике и применять в полевых условиях.
За единицу активности антигемофилического препарата принимается то количество фактора VIII, которое содержится в 1 мл «усредненной» донорской плазмы, т. е. плазмы со 100%-ным содержанием антигемофильного глобулина.
Для купирования кровоизлияний в суставы и небольших кровотечений, в том числе и их предотвращения при удалении зубов, обычно достаточно повысить уровень фактора VIII до 15-20%. Более опасные внутренние и наружные кровотечения, а также развитие гематом в мягких тканях требуют поддержания уровня фактора VIII выше 30-40%, для чего вводят криопреципитат или другие концентраты фактора VIII по 20-30 ЕД/кг; при больших операциях и травмах, гематурии и желудочно-кишечных кровотечениях дозу криопреципитата увеличивают до 40-60 ЕД/кг, а в отдельных случаях - больше.
Вместе с тем избыточные введения криопреципитата нежелательны, так как создают высокую концентрацию фибриногена в крови, вследствие чего нарушается микроциркуляция в органах и возникает опасность тромбозов и ДВС-синдрома.
Частота введения антигемофильных препаратов определяется тем, насколько при каждом введении удалось повысить концентрацию фактора VIII в плазме. Так, если концентрация фактора была повышена до 40%, то уже через 6-8 ч она снизится до 20%, а при начальном повышении до 120% уровень 20% будет достигнут лишь через сутки. Современные концентрированные препараты фактора VIII (криопреципитат и др.) позволяют ограничиться 1-2 внутривенными введениями в сутки. Достаточный эффект заместительной терапии достигается только при соблюдении следующих условий: все антигемофилические препараты вводят внутривенно только струйно, в возможно более концентрированном виде и возможно быстрее после их расконсервирования без смешивания с другими инфузионными растворами. Одна из главных причин неудач заместительной терапии - капельное введение препаратов крови, не повышающих уровня фактора VIII в плазме.
До стойкой остановки кровотечения следует избегать введения любых кровезаменителей и гемопрепаратов (препаратов крови), не содержащих антигемофилических факторов, так как это приводит к разведению фактора VIII и снижению его концентрации.
Раннее удаление (аспирация) излившейся в сустав крови не только сразу же купирует болевой синдром, предотвращает дальнейшее свертывание крови в суставе, но и уменьшает угрозу развития и быстрого прогрессирования остеоартроза. Для предупреждения и купирования вторичных воспалительных изменений после аспирации крови врач назначает введение в сустав 40-60 мг гидроко-тизона. Поддерживающая трансфузионная терапия, проводимая в течение первых 36 дней, предотвращает дальнейшее кровотечение и позволяет рано начать занятия лечебной физкультурой, что способствует более быстрому и полному восстановлению функции пораженной конечности, предотвращает атрофию мышц.
Движения в пораженном суставе следует разрабатывать поэтапно: в первые 5-7 дней после снятия иммобилизирующей повязки выполняют активные движения как в пораженном суставе, так и в других суставах конечности, постепенно увеличивая частоту и длительность упражнений. В дальнейшем с 6-9-го дня переходят на нагрузочные упражнения, пользуясь велоэргометрами, педальными воротами для рук, эластическими тягами. И только на 11-13-й день с целью устранения остаточной тугоподвижности и ограничений максимального сгибания или разгибания с осторожностью под контролем переливаний антигемофильной плазмы или небольших доз криопреципитата выполняют пассивные нагрузочные упражнения.
Одновременно с 5-7-го дня назначают физиотерапевтические воздействия -электрофорез гидрокортизона, анодную гальванизацию.
При кровоизлияниях в мягкие ткани необходима более интенсивная, чем при кровоизлияниях в суставы, терапия антигемофилическими препаратами. При анемизации больного дополнительно назначают переливания эритроцитной массы. Если возникают признаки инфицирования гематомы, то немедленно назначают антибиотики широкого спектра действия. Любые внутримышечные инъекции при гемофилии противопоказаны, так как могут стать причиной обширных гематом и псевдоопухолей. Пенициллин и его полусинтетические аналоги также нежелательны, поскольку в больших дозах вызывают дисфункцию тромбоцитов и усиливают кровоточивость.
Ранняя и интенсивная терапия антигемофилическими препаратами способствует быстрому обратному развитию гематом. Пункций гематом и аспирации из них крови следует избегать. Продолжают трансфузионную терапию от 5-7 до 14 дней. Осумковавшиеся гематомы удаляют, если это возможно, хирургическим путем вместе с капсулой под прикрытием интенсивной терапии концентратами антигемофилических факторов.
При носовых кровотечениях врачу не следует прибегать к тугой тампонаде, особенно задней, так как непосредственно после удаления тампонов кровотечение у таких больных практически всегда возобновляется с еще большей силой.
Для как можно более быстрой остановки носового кровотечения необходимо применять антигемофильную плазму, а также антигемофилические препараты в комбинации с орошением слизистой оболочки носа раствором аминокапроновой кислоты, тромбина или адроксоном.
Серьезную опасность для больных представляют почечные кровотечения, при которых неэффективны переливания антигемофильной плазмы и малых доз криопреципитата. Рекомендуемые средние дозы антигемофилических препаратов (30-40 ЕД/кг) также не всегда купируют эти кровотечения либо останавливают их максимум на 1-2 дня. Усиливает эффективность применения антигемофилических препаратов преднизолон (20-30 мг/сут для взрослых больных).
Для купирования желудочно-кишечных кровотечений следует пользоваться большими дозами концентратов антигемофилических факторов совместно с аминокапроновой кислотой дозировкой до 0,2 г/кг.
Стоит отметить, что использования преднизолона при кровотечениях в желудочно-кишечном тракте необходимо избегать, наиболее опасно применение преднизолона при язвенной болезни как желудка, так и двенадцатиперстной кишки.
Следует помнить, что желудочные кровотечения часто провоцируются приемом в связи с болями в суставах, зубной или головной болью ацетилсалициловой кислоты, бруфена, индометацина, бутазолидонов. У больных гемофилией даже однократный прием ацетилсалициловой кислоты может вызвать желудочное кровотечение.
В профилактике и лечении хронических остеоартрозов и других поражений опорно-двигательного аппарата следует предусматривать различные способы защиты суставов и предупреждения травм конечностей. Для этого в одежду вшивают поролоновые щитки вокруг коленных, голеностопных и локтевых суставов, запрещают все виды спорта, связанные с прыжками, падениями и ушибами (в том числе езду на велосипеде и мотоцикле). Немаловажную роль играют как можно более раннее и полноценное лечение острых кровоизлияний в мышцы и суставы, интенсивная круглогодичная лечебная физкультура. Для этого составляют комплексы из атравматических упражнений в воде, на мягких матах и нагрузочных аппаратах - велоэргометрах, ручных воротах. Занятия нужно начинать в дошкольном или младшем школьном возрасте, т. е. до того, как развились хронические остеоартрозы, нарушения подвижности и другие тяжелые нарушения опорно-двигательного аппарата.
Комплексное лечение дополняют физиотерапевтическими (токи высокой частоты, электрофорез глюкокортикостероидов) и бальнеологическими методами терапии, к которым относятся: грязелечение, рапные и радоновые ванны. При частых и упорно повторяющихся кровоизлияниях в одни и те же суставы методами выбора являются рентгенотерапия и синовэктомия (удаление синовиальной оболочки сустава).
Рентгенотерапию проводят при разовой дозе от 25-50 Р (при острых кровоизлияниях) до 50-100 Р при хроническом остеоартрозе. Сеансы повторяют через 1-2 дня, суммарная доза колеблется от 400 до 1000 Р. У детей моложе 14 лет необходима определенная осторожность из-за возможности повреждения зон роста костей, в связи с чем суммарная доза не должна превышать 400 Р. В последние годы применяется и внутреннее облучение путем введения в суставы радиоактивных изотопов.
Синовэктомия, выполняемая через один разрез, - высокоэффективный метод лечения гемофилических поражений суставов, предупреждения тяжелых остеоартрозов. Данный тип лечения устраняет последующие кровоизлияния в прооперированный сустав, гарантирует сохранение его нормальной конфигурации и функции. Такой эффект выражен при сравнительно раннем выполнении операции - при артрозе I - II степеней, а при поражениях III - IV степеней синовэктомия, как правило, уже нецелесообразна. Как и все другие хирургические вмешательства, синовэктомия выполняется на фоне применения криопреципитата или других концентратов антигемофилических факторов.
Из других ортопедических вмешательств чаще выполняют ахиллопластику, удлинение костей с целью восстановления суставной щели, в запущенных случаях - замыкание суставов компрессией в физиологическом положении. Неоценимую услугу при этом оказывают аппараты Волкова - Оганесяна, Илизарова и других авторов, позволяющие надежно и быстро получить коррекцию без чрезвычайно опасной для больных длительной иммобилизации конечностей. Восстановление суставной щели без использования этих аппаратов вообще практически невозможно.
При достаточной заместительной терапии криопреципитатом (в первые 7-8 дней - 30-40 ЕД/(кг Ч сут), затем - вдвое меньшие количества) у больных обеспечивается вполне надежный гемостаз; кровотечения и кровоизлияния в местах проведения спиц не наблюдаются.
Современные методы заместительной терапии и использование ортопедических аппаратов коренным образом изменили прогноз при переломах костей у больных гемофилией. Если еще недавно такие переломы плохо заживали, часто осложнялись ложными суставами и даже вели к потере конечности, то под влиянием больших доз криопреципитата при одновременном сближении и хорошей фиксации костей с помощью упомянутых выше аппаратов обеспечивается заживление переломов в обычные сроки.
К нерешенным терапевтическим проблемам относится борьба с остеопорозом, внутрикостными кистами и псевдоопухолями, не имеющими тенденции к осумкованию. Эти процессы дают тяжелые осложнения и иногда заставляют прибегать к ампутации конечности.
Лечение осложненных форм гемофилии . Наиболее опасным является обнаружение в крови больных с гемофилией большого количества антител против фактора VIII, которые определяют трансформацию гемофилии в ингибиторную форму. Ингибитор способен инактивировать очень большие количества вводимого извне фактора VIII, в связи с чем основной метод лечения - заместительная трансфузионная терапия - становится малодейственным или совсем неэффективным.
В процессе трансфузионной терапии (с 4-6-го дня) титр антител может резко возрасти.
Если заместительная терапия нужна по жизненным показаниям, то временно преодолеть действие антител удается введением огромных количеств концентрата фактора VIII (по 500-1000 ЕД/кг) или плазмаферезом (удаление у больного нескольких литров плазмы с заменой ее свежей антигемофильной) вместе с введениями мегадоз фактора VIII.
Более перспективным оказалось применение при ингибиторной форме гемофилии А так называемого обходного лечения - введения концентратов факторов IX, X и II. Их применение в средних дозах обеспечивает у половины больных ингибиторной гемофилией остановку кровотечения. Вместе с тем известна тромбогенность этих препаратов, их способность провоцировать развитие ДВС-синдрома и тромбозов, особенно при одновременном применении аминокапроновой кислоты и других гемостатиков. Могут развиться такие нарушения и при обходной терапии ингибиторной формы гемофилии. Так, например, описаны случаи развития инфаркта миокарда у молодых больных гемофилией при повторном применении концентратов факторов протромбинового комплекса. По данным ряда авторов, при ингибиторной гемофилии эффективнее не обычные, а так называемые активированные препараты протромбинового комплекса или комплекса фактора IX. Однако они в 10 и более раз дороже неактивированных концентратов этих же факторов. Для уменьшения опасности развития ДВС-синдрома и тромбозов рекомендуется вводить эти факторы с антитромбином III или с предварительно глубоко замороженной плазмой вместе с небольшими дозами контрикала.
Данные о влиянии преднизолона на количество антител в крови противоречивы, но большинство авторов все же отмечают, что у больных гемофилией такое лечение редко бывает эффективным. Иммунодепрессанты (азатиоприн и др.) чаще снижают количество антител, но их применение опасно из-за развития тромбоцитопении, которая при гемофилии может существенно усугубить геморрагические явления.
Если у больных с ингибиторной формой гемофилии отсутствует положительная динамика, то при проведении заместительной терапии перед хирургическими вмешательствами всегда нужно тщательно проверять, присутствует ли указанный ингибитор в плазме.
При обострении вторичного ревматоидного синдрома, а также при проведении курсов интенсивной заместительной терапии, особенно если она не ослабляет, а усиливает боли в суставах, хороший эффект дает преднизолон (20-40 мг в день в течение месяца с последующим постепенным снижением дозы до минимума).
Большое значение в профилактике кровотечений имеет сведение к минимуму с раннего детского возраста опасности травм, порезов и т. д. Из обихода исключают легко ломающиеся игрушки (в том числе металлические и пластмассовые), а также неустойчивые и тяжелые предметы. Мебель должна быть с закругленными гранями, выступающие края обматывают ватой или поролоном, пол покрывают ворсовым ковром. Предпочтительнее общение и игры больных с девочками, а не с мальчиками.
Для больного важен правильный выбор профессии и места работы. В наиболее тяжелых случаях единственным эффективным методом смягчения болезни служит систематическое, один раз в 10 дней, внутривенное введение криопреципитата или любого иного имеющегося в наличии концентрата фактора VIII. Разовая доза препарата при такой профилактике составляет 400-800 ЕД.
Огромное значение для предупреждения тяжелых поражений опорно-двигательного аппарата имеет как можно более раннее введение препаратов фактора VIII при ушибах и других травмах, а также при проявлении острой боли в суставе, говорящей о том, что в него началось подтекание крови. Немедленное введение концентратов антигемофилических факторов (специальной бригадой «Скорой помощи» или гемофилического центра либо прошедшими обучение родителями больного) обрывает образование или обострения кровоизлияний в самом начале, предотвращает деструктивные изменения в суставе. Неотложная доврачебная помощь - важнейший компонент физической реабилитации больных, профилактики тяжелых и необратимых изменений в суставах, костях и мышцах. Она резко уменьшает число госпитализаций, среднегодовую длительность пребывания в больнице, не отрывает детей от учебы.
С интенсификацией заместительной трансфузионной терапии увеличивается число инфицированных вирусом сывороточного гепатита, учащаются тяжелые реакции на введение гемопрепаратов и возникает ряд других иммунных нарушений.
Генопрофилактика гемофилии пока не разработана. Определение пола путем исследования полового хроматина и кариотипа амниотических клеток, полученных методом амниоцентеза, позволяет своевременно прервать беременность, но не показывает, является ли плод носителем гемофилического гена. Беременность сохраняют, если плод мужского пола, так как все сыновья больных рождаются здоровыми, и прерывают, если плод женского пола, поскольку все дочери больных гемофилией являются носителями заболевания.
У женщин - носителей гемофилии, имеющих 50%-ную вероятность родить больного (если плод мужского пола) или передатчицу гемофилии (если плод женского пола), рождение только девочек переносит опасность появления в семье больных гемофилией с первого поколения на второе, а также одновременно увеличивает общее число носителей заболевания.
Минздрав РФ одобрило препарат Револейд (Элтромбопаг) к применению у детей. Новый препарат показан пациентам страдающим хронической иммунной тромбоцитопенией (идиопатической тромбоцитопенической пурпурой, ИТП), редким заболеванием системы крови.
03.03.2017
Канадские ученые из университета в Оттаве намерены произвести революцию в восстановительной медицине. В одном из последних экспериментов им удалось вырастить человеческое ухо из обычного яблока.
27.02.2017
Специалисты Первого медицинского университета имени Павлова в Санкт-Петербурге создали наночастицы с помощью которых возможна диагностика инфарктных и пред инфарктных состояний больного. Так же в перспективе исследований, наночастицы будут применяться...
Медицинские статьи
Почти 5% всех злокачественных опухолей составляют саркомы. Они отличаются высокой агрессивностью, быстрым распространением гематогенным путем и склонностью к рецидивам после лечения. Некоторые саркомы развиваются годами, ничем себя не проявляя...
Вирусы не только витают в воздухе, но и могут попадать на поручни, сидения и другие поверхности, при этом сохраняя свою активность. Поэтому в поездках или общественных местах желательно не только исключить общение с окружающими людьми, но и избегать...
Вернуть хорошее зрение и навсегда распрощаться с очками и контактными линзами - мечта многих людей. Сейчас её можно сделать реальностью быстро и безопасно. Новые возможности лазерной коррекции зрения открывает полностью бесконтактная методика Фемто-ЛАСИК.
Косметические препараты, предназначенные ухаживать за нашей кожей и волосами, на самом деле могут оказаться не столь безопасными, как мы думаем
Ключевые понятия нарушения гемостаза:
1. Нарушения гемостаза, коагулопатия
(coagulopathia, коагуло- + греч. patos – страдание,болезнь) – нарушение функции свёртывающей и противосвёртывающей систем крови.
2. Гиперкоагуляционно-тромботическое состояние
– состояние, сопровождающееся патологическим процессом усиления свёртываемости крови вследствие повышенной агрегации тромбоцитов, активации плазменных и тканевых факторов свёртывания крови с образованием тромбоцитарного и фибринового сгустков.
3. Гипокоагуляционно-геморрагическое состояние
– состояние, сопровождающееся патологическим процессом уменьшения свёртываемости крови вследствие снижения агрегации тромбоцитов, инактивации плазменных и тканевых факторов свёртывания крови с возникновением кровоточивости и кровотечений.
4. ДВС-синдром (тромбо-геморрагическое состояние)
– синдром диссеминированного внутрисосудистого свёртывания крови – типический патологический процесс нарушения гемостаза в результате последовательно протекающих реакций свёртывания крови: гиперкоагуляции (образование диссеминированных тромбов в микроциркуляторной сосудистой сети) и гипокоагуляции (истощение тромбогенных факторов и усиление фибринолиза), сопровождающихся массивным кровотечением, тяжёлым гемокоагуляционным шоком и острой дистрофией внутренних органов.
При каких заболеваниях возникает нарушение гемостаза:
Гемостаз - это сложный процесс, который предотвращает или останавливает истечение крови из просвета сосуда, обеспечивает возникновение свертка фибрина, необходимого для восстановления целостности ткани, и, наконец, удаляет фибрин, когда нужда в нем отпадает. В этом процессе участвуют четыре основных физиологических механизма.
С помощью системы гемостаза кровь выполняет свою важнейшую функцию - поддержание жидкого состояния крови, протекающей в кровеносных сосудах, и свертывание крови при нарушении целостности сосудистой стенки и, тем самым, прекращение кровотечения и сохранение объема и состава крови. Система гемостаза многокомпонентна. В ней участвуют тромбоциты и другие клетки крови, сосудистая стенка, экстраваскулярная ткань, биологически активные вещества (тромбоцитарно-сосудистый гемостаз), плазменные, тканевые факторы свертывания крови (коагуляционный гемостаз), находящиеся в тесном взаимодействии с противосвертывающей, фибринолитической и калликреин-кининовой системами. Нарушение любого из этих компонентов ведет к патологии гемостаза.
Классификация нарушения гемостаза. Патология гемостаза классифицируется по преимущественному поражению различных его компонентов на нарушения тромбоцитарно-сосудистого гемостаза и коагуляционного гемостаза. По этиологии эти нарушения могут быть приобретенными и наследственными, а по направленности изменений подразделяться на понижение свертывания крови (гипокоагуляцию) и повышение свертывания крови (гиперкоагуляцию), которое может быть локальным (тромбоз) и генерализованным (ДВС-синдром).
Понижение свертывания крови
Понижение свертывания крови проявляется повышенной кровоточивостью (геморрагическим синдромом) - повторными кровотечениями, кровоизлияниями, возникающими как самопроизвольно, так и при незначительных травмах.
Тромбоцитарно-сосудистый гемостаз нарушается при количественных и качественных изменениях тромбоцитов (тромбоцитопениях и тромбоцитопатиях), а также поражениях сосудистой стенки.
Тромбоцитопенией называется уменьшение содержания тромбоцитов в крови ниже нормы (180-320 Г/л или 180-320x109/л). Однако спонтанные кровотечения возникают лишь при снижении их числа меньше 30 Г/л. Под тромбоцитопатиями понимают качественную неполноценность и дисфункцию тромбоцитов при нормальном или пониженном их содержании.
Причины пониженного свертывания крови. Причиной возникновения тромбоцитопении нередко являются иммунные реакции при изменении антигенной структуры тромбоцитов под действием вирусов, лекарственных препаратов, выработке антитромбоцитарных аутоантител (при хроническом лимфолейкозе, идиопатической тромбоцитопении), несовместимости тромбоцитарных антигенов матери и плода. Кроме того, тромбоцитопения развивается вследствие поражения мегакариоцитарного ростка костного мозга ионизирующей радиацией, химическими веществами или вытеснения его опухолевыми метастазами, лейкозными инфильтратами. Снижение тромбоцитопоэза может быть обусловлено дефицитом цианокобаламина и фолиевой кислоты, наследственным дефектом образования тромбоцитов (в том числе при дефиците тромбоцитопоэтинов). Тромбоцитопения возникает в результате механического повреждения тромбоцитов при спленомегалии, искусственных клапанах сердца, а также усиленного потребления тромбоцитов при локальном и генерализованном внутрисосудистом свертывании крови.
К этиологическим факторам, вызывающим тромбоцитопатию, относятся действие токсических веществ и лекарственных препаратов (алкоголь, ацетилсалициловая кислота), ионизирующей радиации, эндогенных метаболитов (при уремии, циррозе печени); дефицит цианокобаламина, гормональные нарушения (гипотиреоз). Наблюдаются и генетические дефекты структуры мембраны и биохимического состава тромбоцитов (дефицит тромбостенина, фактора 3, АТФ, АДФ, Г-6-ФДГ, мембранных рецепторов для факторов V, VIII, XI и др.).
При геморрагических вазопатиях поражение сосудистой стенки, приводящее к нарушению тромбоцитарно-сосудистого гемостаза и кровоточивости, возникает вследствие повышения проницаемости стенки кровеносных сосудов и ее деструкции при нарушении синтеза коллагена (при алиментарном дефиците аскорбиновой кислоты, наследственных дефектах синтеза коллагена), при действии биологически активных веществ (аллергия), радиотоксинов (лучевая болезнь), иммунных геморрагических васкулитах, снижении ангиотрофической функции тромбоцитов при тромбоцитопениях и тромбоцитопатиях, разрушении сосудистой стенки лейкозными инфильтратами. Одной из причин кровоточивости является уменьшение выработки эндотелием сосудистой стенки фактора Виллебранда - крупномолекулярного компонента VIII фактора свертывания-крови (наследственная болезнь Виллебранда). Этот фактор накапливается в тромбоцитах и освобождается при их дегрануляции.
Он необходим для нормальной адгезии тромбоцитов к коллагену стенки, и без него не формируется тромбоцитарный тромб. Геморрагический синдром наблюдается и при усилении перекисного окисления мембранных фосфолипидов, в результате чего в эндотелии синтезируется и секретируется избыточное количество мощных ингибиторов агрегации тромбоцитов - простациклинов. Кроме того, к снижению тромбоцитарно-сосудистого гемостаза приводит нарушение нейрогенной и гуморальной регуляции сосудистого тонуса, понижение которого ведет к невозможности закупорки мелких сосудов тромбоцитарным тромбом.
Патогенез пониженного свертывания крови. Выделяют четыре основных механизма возникновения тромбоцитопений: уменьшение продукции, усиленное разрушение, повышенное потребление (тромбообразование), перераспределение тромбоцитов.
Нарушение гемостаза и развитие кровоточивости при тромбоцитопении обусловлено следующими механизмами:
- повышением проницаемости микрососудов для эритроцитов и других составных частей крови (диапедезная геморрагия) и ломкости сосудов вследствие дистрофии стенки при выключении ангиотрофической функции тромбоцитов;
- уменьшением адгезивно-агрегационной функции тромбоцитов;
- нарушением реакции освобождения тромбоцитарных факторов свертывания крови, АДФ, серотонина, адреналина, антигепаринового фактора, следствием чего является недостаточное формирование тромбоцитарного тромба, отсутствие спазма сосудов и замедление свертывания;
- уменьшением ретракции сгустка в результате снижения активности сократительного белка тромбоцитов - тромбостенина (фактор 8 тромбоцитов).
В патогенезе тромбоцитопатий можно выделить два основных механизма их возникновения - продукция патологических тромбоцитов в костном мозге и деструкция тромбоцитов во всех отделах системы крови. Патогенез нарушения тромбоцитарно-сосудистого гемостаза при тромбоцитопатиях такой же, как и при тромбоцитопениях, так как связан с выключением функций тромбоцитов.
Нарушение коагуляционного гемостаза, приводящее к развитию кровоточивости, может быть вызвано следующими факторами:
- приобретенным и наследственным уменьшением или извращением синтеза плазменных и тромбоцитарных факторов свертывания крови и компонентов калликреин-кининовой системы;
- ингибированием или повышенным потреблением этих факторов;
- увеличением эндогенных антикоагулянтов;
- активизацией фибринолитической системы;
- передозировкой антикоагулянтов, фибринолитических и дефибринирующих препаратов. Все это лежит в основе нарушения одной из трех фаз свертывания крови и ретракции сгустка или же сочетанного их изменения.
Причинами нарушения первой фазы свертывания крови - образования тромбопластина - является снижение продукции факторов (IX, X) при патологии печени, образование антител к некоторым факторам (VIII, IX) при заболеваниях, в патогенезе которых имеется аутоиммунный компонент (лейкозы, коллагенозы), или же передозировка такого универсального антикоагулянта, как гепарин. Известны генетические дефекты синтеза VIII, IX и XI факторов, дефицит которых лежит в основе развития гемофилии (соответственно последовательности этих факторов - гемофилии А, В и С).
Нарушение второй фазы свертывания крови - образования тромбина - возникает не только при заболеваниях печени, но и при гипо- и авитаминозах К, когда тоже понижается синтез в печени факторов II, V, VII, участвующих в этой фазе (при механической желтухе, энтерите, обширной резекции тонкой кишки, лекарственном дисбактериозе). Возможно появление иммунных ингибиторов факторов V, VII (например, при лечении стрептомицина сульфатом), усиленное их выведение почками, наследственный дефицит (фактора V при парагемофилии) или же инактивация компонентами противосвертывающей системы - антитромбинами, гепарином (при анафилактическом шоке, передозировке гепарина).
Геморрагический диатез, связанный с нарушением третьей фазы свертывания - фазы образования фибрина, возникает при уменьшении синтеза фибриногена в пораженных патологическим процессом печени, легких или же в результате наследственной гипо-, афибриногенемии и дефицита фибринстабилизирующего фактора (фактора XIII). Однако значительно чаще нарушение третьей фазы является следствием усиления фибринолиза при травме (операции) легких, матки, поджелудочной железы; ожоге, шоке. Это обусловлено повышенным поступлением в кровь активаторов профибринолизина (плазминогена) - тканевых, микробных фибринокиназ, лейко- и эритроцитарных активаторов, компонентов калликреин-кининовой системы и системы комплемента комплексов гепарина с фибриногеном, профибринолизином и адреналином (эти комплексы обеспечивают неферментативный фибринолиз, по Б. А. Кудряшову).
Патогенез. Главными звеньями в патогенезе геморрагического диатеза, развившегося при нарушении любой из фаз свертывания крови, являются хроническая кровопотеря и ее последствия, а также структурные и функциональные изменения в месте кровоизлияний (в суставах, внутренних органах, коже и других тканях).
Повышение свертывания крови
Повышение свертывания крови проявляется локальным (тромбоз) или генерализованным внутрисосудистым свертыванием крови, в основе чего лежит нарушение тромбоцитарно-сосудистого и коагуляционного гемостаза.
Гиперкоагуляция может быть обусловлена:
- повышением функциональной активности системы свертывания крови за счет увеличенного - - поступления в кровь прокоагулянтов и активаторов свертывания крови;
- увеличением в крови содержания тромбоцитов;
- снижением антитромботических свойств сосудистой стенки;
- уменьшением активности противосвертывающей системы крови;
- ослаблением фибринолиза.
Генерализованное (диссеминированное) внутрисосудистое свертывание крови (ДВС-синдром) - тяжелое нарушение гемостаза, возникающее при избыточном поступлении в кровь прокоагулянтов и активаторов свертывания крови, что ведет к образованию множественных микротромбов в сосудах микроциркуляторного русла, а затем развитию гипокоагуляции, тромбоцитопении и геморрагии в результате "потребления" факторов системы свертывания и повышения функциональной активности системы противосвертывания и фибринолиза крови с последующим истощением всех трех систем.
Этиология. Универсальность и неспецифичность ДВС-синдрома обусловлены многообразием причинных факторов его возникновения. К ним относятся прежде всего генерализованные инфекции и септические состояния, все виды шока, травматические хирургические операции, акушерская патология (преждевременная отслойка, ручное отделение плаценты), острый внутрисосудистый гемолиз, уремия при почечной недостаточности, все терминальные состояния.
Патогенез. Главным звеном в патогенезе генерализованной гиперкоагуляции является нарушение баланса между калликреин-кининовой, свертывающей, противосвертывающей и фибринолитической системами крови при поступлении в сосудистое русло большого количества прокоагулянтов и их активаторов. Это и приводит к нарушению столь важной функции крови, как сохранение ее нормального агрегатного состояния, в результате чего в фазу гиперкоагуляции кровь в сосудах свертывается и прекращается ее циркуляция с развитием тяжелых дистрофических и функциональных нарушений в органах и тканях, нередко несовместимых с жизнью. В последующую фазу гипокоагуляции разжижение крови и потеря способности к свертыванию и агрегации тромбоцитов вызывают кровотечение, которое плохо поддается терапевтической коррекции. При благоприятном исходе наступает третья - восстановительная фаза, при которой происходит нормализация гемостаза.
У больного цингой наблюдаются кровоточивость десен и петехии на коже. Что вызывает нарушение гемостаза при этом заболевании?
A. @ Нарушение синтеза коллагена
B. Тромбоцитопения
C. Избыток антикоагулянтов
D. Активация фибринолиза
E. Дефицит прокоагулянтов
У мальчика с геморрагическим синдромом в крови отсутствует антигемофильный глобулин А (фактор VIII). Недостаточность какого механизма гемокоагуляции имеет место у этого больного?
@ Внутренний механизм формирования протромбиназы
Превращение фибриногена в фибрин
Внешний механизм формирования протромбиназы
Превращение протромбина в тромбин
Ретракция кровяного сгустка
Мужчина который в течение двух лет болеет хроническим миелоидным лейкозом, поступил в больницу в состоянии острой почечной недостаточности. Что может быть причиной острой почечной недостаточности у этого больного?
@ ДВС – синдром
Лимфопения
Нейтропения
Тромбоцитопения
Недостаточная активность каких факторов свертывания крови обусловливает развитие геморрагического синдрома у больных гиповитаминозом К?
@ X, IX, VII, II
Фактор Виллебранда
У пациентки, которая систематически употребляет ацетилсалициловую кислоту, появилось кровоизлияние. С уменьшением активности каких ферментов тромбоцитов связано развитие тромбоцитопатии в данном случае?
@ Циклооксигеназы
Липооксигеназы
Пероксидазы
Цитохромоксидазы
Глюкозо–6–фосфатдегидрогеназы
У девочки периодически возникают кровотечения из носа, появляются небольшие геморрагические высыпания на коже. При обследовании обнаружено: время кровотечения – 10 минут, снижена адгезивная способность тромбоцитов и низкая активность ф. VIII (VIII:С). Какое заболевание у ребенка?
@ Болезнь Виллебранда
Гемофилия А
Гемофилия В
Наследственная дисфибриногенемия
Тромбоцитопения
После перенесенной ангины у девочки 5 лет наблюдаются петехиальная сыпь на коже туловища и конечностей, кровотечения из десен. При обследовании обнаружено уменьшение количества тромбоцитов в крови. При каком уровне тромбоцитопении (Г/л) появляются ее клинические признаки?
Что может быть главным звеном в патогенезе тромбофилии в пациентки, которая страдает тромбофлебитом вен нижних конечностей?
@ Недостаточность антикоагулянтов
Тромбоцитопения
Тромбоцитопатия
Недостаточность прокоагулянтов
С целью диагностики какого сдвига в системе гемокоагуляции назначают исследование уровня продуктов деградации фибрина в крови?
Тромбоцитопатия
Тромбоцитопения
Вазопатия геморрагическая
У больного диагностирован генетический дефект мембранного рецептора к фактору Виллебранда – гликопротеиду (ГП) Ib, который отвечает за начальный этап адгезии тромбоцитов к коллагену. Как называется эта болезнь?
@ Бернара – Сулье
Виллебранда
Аддисона – Бирмера
Вильсона – Коновалова
Гланцмана – Негели
У больного диагностирован наследственный дефект ГП IIb–IIIa – мембранного рецептора, который обеспечивает соединение фибрина с мембраной тромбоцитов и нужен для их агрегации. О какой болезни идет речь?
@ Гланцмана – Негели
Виллебранда
Аддисона – Бирмера
Вильсона – Коновалова
Бернара – Сулье
У ребенка с комбинированным иммунодефицитом при обследовании крови обнаружено уменьшение адгезии тромбоцитов к коллагену, их агрегации, ослабление коагуляции крови и ретракции кровяного сгустка. При каком иммунодефиците наблюдаются подобные изменения?
@ Вискотта – Олдрича
Швейцарского типа
Ди – Джорджи
Незелофа
У больной девочки диагностирована тромбастения Гланцманa. Какое нарушение в системе гемостаза имеет место при этом?
@ Дисагрегационная тромбоцитопатия
Абсолютная тромбоцитопения
Дисадгезивная тромбоцитопатия
Дефицитная тромбоцитопатия
Дисдегрануляционная тромбоцитопатия
Участники экспедиции, которые находились на севере, жалуются на кровоточивость десен и петехиальные кровоизлияния на коже. Из анамнеза известно – в рационе не хватало аскорбиновой кислоты, и это привело к ломкости сосудистой стенки. Какая это по патогенезу вазопатия?
@ Диспластическая
Воспалительная
Метапластическая
Дистрофическая
Иммунная
У больного после длительной операции на поджелудочной железе, послеоперационная рана долго кровоточила. По данным коагулограммы обнаружено существенное повышение уровня плазмина. Какая по патогенезу коагулопатия наблюдается в данном случае?
@ Фибринопатия
Протромбиназопатия
Тромбопластинопатия
Тромбинопатия
Вазопатия
У больного диагностирован генетический дефект V фактора, который становится нечувствительным к инактивации антитромботическим комплексом тромбомодулин–протеин С, что снижает способность сосудистой стенки ограничивать образование фибрина. Какая патология свертывания крови возникнет при этой аномалии?
@ Тромбофилия
Тромбоцитопения
Тромбоцитопатия
Геморрагический синдром

ПАТОФИЗИОЛОГИЯ СИСТЕМЫ ГЕМОСТАЗА
Значение системы гемостаза
1. Сохранение крови в жидком состоянии (адекватное соотношение активности свертывающей и противосвертывающей систем)
2. Предупреждение и остановка кровотечения (поддержание постоянного объема циркулирующей крови)

ВИДЫ ГЕМОСТАЗА
СОСУДИСТО-ТРОМБОЦИТАРНЫЙ
(ПЕРВИЧНЫЙ)
- ОСТАНОВКА КРОВОТЕЧЕНИЯ В МИКРОСОСУДАХ
КОАГУЛЯЦИОННЫЙ
(ВТОРИЧНЫЙ)
ФОРМИРОВАНИЕ ФИБРИНОВЫХ СГУСТКОВ

КОМПОНЕНТЫ СИСТЕМЫ ГЕМОСТАЗА
* СОСУДИСТАЯ СТЕНКА
* ТРОМБОЦИТЫ (моноциты, эритроциты)
* ПЛАЗМЕННЫЕ СИСТЕМЫ :
- ПРОКОАГУЛЯНТЫ
- АНТИКОАГУЛЯНТЫ
- ФИБРИНОЛИТИЧЕСКАЯ
- КАЛЛИКРЕИН-КИНИНОВАЯ

КЛАССИФИКАЦИЯ НАРУШЕНИЙ ГЕМОСТАЗА
ЗА ЭТИОЛОГИЕЙ
- НАСЛЕДСТВЕННЫЕ
- ПРИОБРЕТЕННЫЕ
ЗА МЕХАНИЗМОМ РАЗВИТИЯ
- НАРУШЕНИЯ СОСУДИСТО-ТРОМБОЦИТАРНОГО
ГЕМОСТАЗА
- НАРУШЕНИЯ КОАГУЛЯЦИОННОГО ГЕМОСТАЗА
ЗА НАПРАВЛЕННОСТЬЮ ИЗМЕНЕНИЙ
- ГИПОКОАГУЛЯЦИЯ
- ГИПЕРКОАГУЛЯЦИЯ

ГИПОКОАГУЛЯЦИЯ
Снижение способности крови сворачиваться с появлением склонности к повторным кровотечениям и кровоизлия-ниям (спонтанным или после незначительных травм)

ЭТИОЛОГИЯ
1. ТРОМБОЦИТОПЕНИЯ
2. ТРОМБОЦИТОПАТИЯ
3. ВАЗОПАТИЯ
4. КОАГУЛОПАТИЯ

ТРОМБОЦИТОПЕНИЯ
Патологическое состояние которое характеризуется понижением содержания тромбоцитов в крови (меньше 150·109 /л)

НАСЛЕДСТВЕННАЯ ТРОМБОЦИТОПЕНИЯ
Как правило, одновремен-но сопровождается врож-деннными дефектами тромбоцитов

ПРИОБРЕТЕННАЯ ТРОМБОЦИТОПЕНИЯ (КЛАССИФИКАЦИЯ ЗА МЕХАНИЗМОМ РАЗВИТИЯ)
ПОВРЕЖДЕНИЕ ТРОМБОЦИТОВ
- имунными комплексами
- механическая травматизация (спленомегалия, гемангиома)
УГНЕТЕНИЕ ОБРАЗОВАНИЯ ТРОМБОЦИТОВ
(апластическая анемия, химическое и радиационное повреждение красного костного мозга, замещение кроветворной ткани опухолью)
ПОВЫШЕННОЕ ИСПОЛЬЗОВАНИЕ ТРОМБОЦИТОВ
(тромбоз, ДВС-синдром)

ИММУННАЯ ТРОМБОЦИТОПЕНИЯ
ГЕТЕРОИММУННАЯ
* Возникает чаще у детей
** Причина – изменение антигенной структуры тромбоцитов (при оседании вирусов краснухи, оспы, аденовирусов; гаптенов медикаментозного происхождения – хинидин, сульфаниламиды, рифампицин; вакцины)
***Течение благоприятное (при устранении причины наступает полное выздоровление)

ИММУННАЯ ТРОМБОЦИТОПЕНИЯ
АУТОИММУННАЯ
Возникает чаще у взрослых
Причина – отсутствие иммунной толерант-ности к антигенам собственных тромбоцитов
Провоцирующие факторы: лекарства, вирусы, бактерии

Аутоиммунная тромбоцитопения
БОЛЕЗНЬ ВЕРЛЬГОФА
(аутоиммунная хроническая тромбоцитопеническая пурпура)
* На поверхности тромбоцитов количество Ig G увеличивается в 10 раз
* Основным местом синтеза Ig G является селезенка
* Принцип лечения :
- спленэктомия
- кортикостероиды
- иммунодепресанты
* Полного излечения не бывает

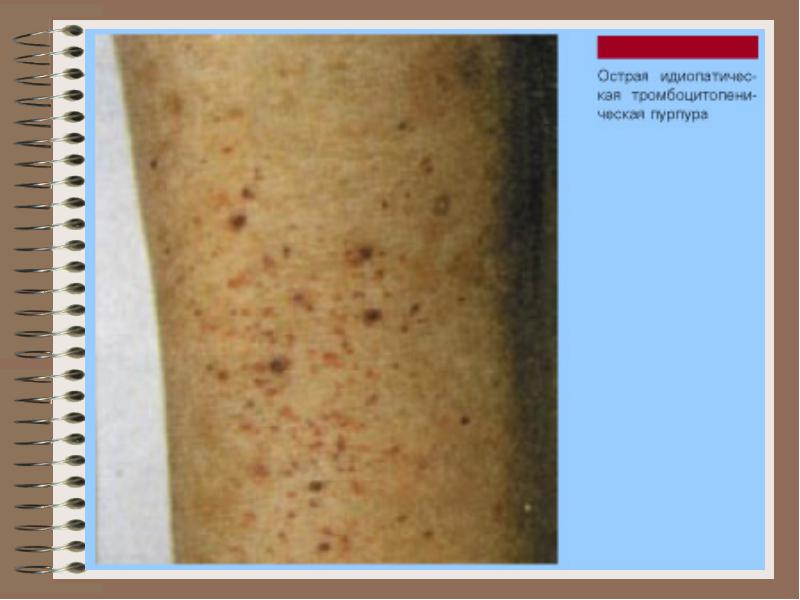
ТРОМБОЦИТОПАТИЯ
Нарушенние гемостаза вследствие качественной неполноценности или дисфункции тромбоцитов, которое характеризуется нарушением сосудисто-тромбоцитарного гемостаза, появлением кровоточи-вости тканей и органов

БЕЗ НАРУШЕНИЯ РЕАКЦИИ ОСВОБОЖДЕНИЯ ГРАНУЛ
Тромбастения Гланцмана
*Наследование - аутосомно-рецессивное
*Причина - отсутствие гликопротеидов 2в и 3а в оболочке тромбоцитов
*Патогенез - тромбоциты не взаимодей-ствуют с фибриногеном и не агрегируют
*Признаки: петехии, носовые кровотече-ния, маточные кровотечения (могут быть смертельными!! )

Наследственная тромбоцитопатия
С НАРУШЕНИЕМ РЕАКЦИИ ОСВОБОЖДЕНИЯ ГРАНУЛ
Наследование - аутосомно-рецессивное
Причина – нарушение активности циклоксигена-зы, низкая активность контрактильных белков
Патогенез – отсутствие агрегации при взаимодействии с коллагеном, отсутствие освобождения гранул
Признаки:

Наследственная тромбоцитопатия
С НАРУШЕНИЕМ НАГРОМОЖДЕНИЯ И ОСВОБОЖДЕНИЯ СОДЕРЖИМОГО ГРАНУЛ
Болезнь Херджманского-Пудлака (АР)
* Причина – нарушение накопления плотных гранул (АДФ, адреналин, серотонин, Са2+)
* Патогенез – отсутствует агрегация при взаимодей-ствии с коллагеном, отсутствует освобождение содержимого гранул
* Признаки: петехии, носовые кровотечения, маточные кровотечения

Наследственная тромбоцитопатия
С НАРУШЕНИЕМ АДГЕЗИИ И АГРЕГАЦИИ ТРОМБОЦИТОВ
Синдром Виллебранда-Юргенса (АР)
Причина – дефицит фактора Виллебранда
Патогенез – нарушена адгезия тромбоцитов вследствие дефицита фактора 8
Болезнь Бернара Сульє (АР)
Причина – отсутствие гликопротеида 1 на тромбоцитах
Патогенез – нарушено взаимодействие тромбоцитов с фактором Виллебранда, ф. 5, ф. 11
Признаки – капиллярные кровотечения (особенно опасны при половом созревании или родах)

Наследственная тромбоцитопатия
ДЕФИЦИТ И СНИЖЕННАЯ ДОСТУПНОСТЬ ф.3
Тромбоцитопатия Боуе и Овена
Причина - дефицит ф.3 тромбоцитов
Патогенез – отсутствует взвємодействие тромбоцитов и прокоагулянтов
Признаки: петехии, носовые кровотечения, маточные кровотечения

Наследственная тромбоцитопатия
Тромбоцитопатии в сочетании сдругими наследственными аномалиями
Синдром Вискотта-Олдриджа
- Причина – в тромбоцитах мало плотных гранул (АДФ, серотонин, адреналин, Са2+), альфа-гранул (бета-тромбоглобулин, фибриноген, фибронектин, ростовой фактор)
- Патогенез –сниженная адгезия и агрегация тромбо-цитов, нарушено освобождение гранул
- Признаки: геморрагический синдром появляєтся рано, могут быть смертельные кровотечения

Приобретенная тромбоцитопатия (Этиология)
1. Лейкозы - мало гранул в тромбоцитах вследствие ускоренного созревания, снижена адгезия и агрегация
2. Нагромождение Ig М – повреждение рецепторов иммунными комплексами, нарушение взаимодействия тромбоцитов с прокоагулянтами (иммунные болезни)
3. Гиповитаминоз В12 – нарушено освобождение гранул
4. Медикаментозные влияния

Медикаментозная тромбоцитопатия
* Ингибиторы синтеза тромбоксана А2
-стероидные противовоспалительные препараты
- нестероидные противовоспалительные препараты (аспирин блокирует агрегацию тромбоцитов на 4-6 дней)
* Стимуляторы образования цАМФ
-папаверин
-эуфиллин
-анаболические стероиды
* Антагонисты ионов Са
-верапамил
-коринфар

ВАЗОПАТИЯ
Геморрагический диатез обусловленный функциональной и морфологической неполноцен-ностью сосудистой стенки
- врожденная
- приобретенная

ВРОДЖЕННАЯ ВАЗОПАТИЯ
Бол. Рандю-Ослера (геморрагическая телеангиоэктазия)
Бол. Фабри (диффузная ангиокера-тома туловища)
Наследственный тромбоцитопени-ческий микроангиоматоз

ВРОДЖЕННАЯ ВАЗОПАТИЯ
Причина – наследственное нарушение развития соединительной ткани, в т.ч. субэндотелия сосудов
Характеристика
- очаговое истончение сосудов
- расширение просвета микрососудов
- мало коллагеновых волокон в субэндотелии
- сосуды легко травмируються
- слабкая адгезия и агрегация тромбоцитов вследствие дефицита коллагеновых волокон
**Признаки – кровотечения носовые, легочно-бронхиальние и желудочно-кишечные (бывают смертельные)

ПРИОБРЕТЕННАЯ ВАЗОПАТИЯ
1. Идиопатическая (саркома Капоши)
- этиология – неизвестна
2. Застойная (дерматит Клотца, дерматит Фавра-Ракушо)
- этиология – хр.сердечная недостаточность, локальная венозная недостаточность
3. Дистрофическая
Стероидная пурпура - гиперфункция надпочечников, лечение кортикостероидами – угнетают синтез коллагена
Скорбут – дефицит вит.С
Бол.Шенляйн-Геноха – повреждение сосудов иммунными комплексами
4. Неврогенная
Клинические признаки – кожные формы кровоточивости

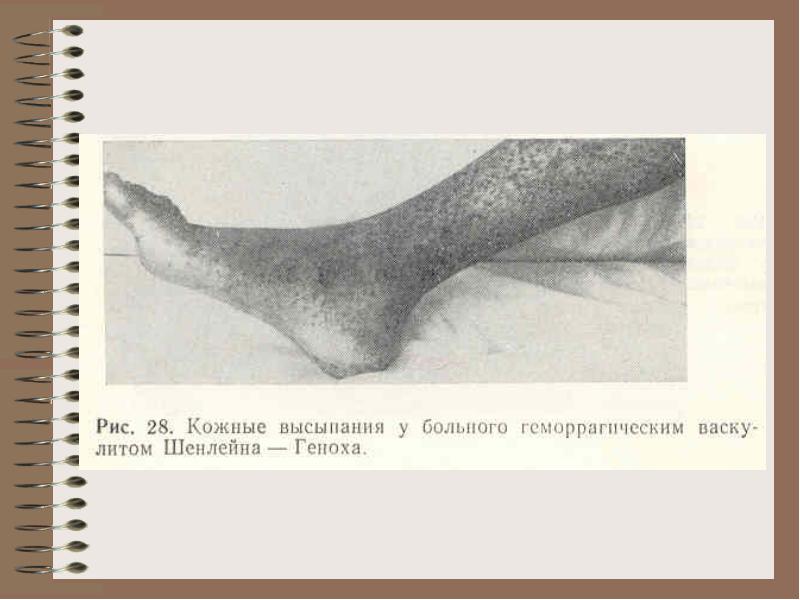
КОАГУЛОПАТИЯ
Геморрагический диатез, который возникает в результате патологии коагуляционной системы гемостаза
** наследственная
** приобретенная

НАСЛЕДСТВЕННАЯ КОАГУЛОПАТИЯ
Генетически обусловленное нарушение свертывания крови вследствие дефицита или молекулярной аномалии веществ, отвечающих за коагуляционный гемостаз

НАСЛЕДСТВЕННАЯ КОАГУЛОПАТИЯ
КЛАССИФИКАЦИЯ
1. Коагулопатия вследствие изолированного нарушения внутреннего механизма формирования протромбиназной активности (гемофилии А, В, С, б.Виллебранда, дефицит Хагемана)
2. Коагулопатия вследствие изолированного нарушения внешнего механизма формирования протромбиназной активности (гипопроконвертинемия – дефицит 7 ф.)
3. Комбинированное нарушение внешнего и внутреннего механизма формирования протромбиназной активности (парагемофилия – дефицит 5 ф., б.Стюарта-Прауэра – дефи-цит 10 ф.)
4. Нарушение конечной стадии свертывания крови (афибриногенемия)

СТАТИСТИКА
Среди всех форм коагулопатий страдают:
Гемофилией А 68 – 78%
Б. Виллебранда 9 – 18 %
Гемофилией В 6 – 13 %
Гемофилией С, парагемофилией и гипопроконвертинемией 1 – 2 %
Остальные формы – клиническая казуистика

Гемофилия А
Геморрагический диатез, обусловленный наслед-ственным дефицитом прокоагулянтной части фактора 8
Фактор 8 (высокомолекулярный белок)
1. Гликопротеин прокоагулянт (VIII:К)
2. Гликопротеин, осуществляющий адгезию тромбоцитов (VIII:ФВ)
3. Гликопротеин, активирующий адгезию тромбоцитов под влиянием ристомицина (VIII:Ркоф)
4. Антигенный маркер VIII:К (VIII:К АГ)
5. Антигенный маркер VIII: Ркоф (VIII: Ркоф АГ)
Активность VIII:К и VIII:ФВ снижается при уменьшении мультимерной структурыи всего 8 фактора

Гемофилия А
* Этиология – аномалия гена в Х-хромосоме, который контролирует синтез прокоагулянтной части ф. 8 (VIII:К)
** Болеют – мужчины (46, XhY)
** Виды
- Гемофилия А+ (антигенположительная форма – синтезируется аномальный VIII:К ), страдают 8 –10 %
- Гемофилия А- (антигенотрицательная форма – не синтезируется VIII:К ), страдают 90 –92 %
**** Клиника: кровоизлияния в большие суставы, гематомы (подкожные, внутримышечные), сильные и длительные посттравматические кровотечения. Возможны кровоизлия-ния в органы брюшной полости, желудочно-кишечные кровотечения

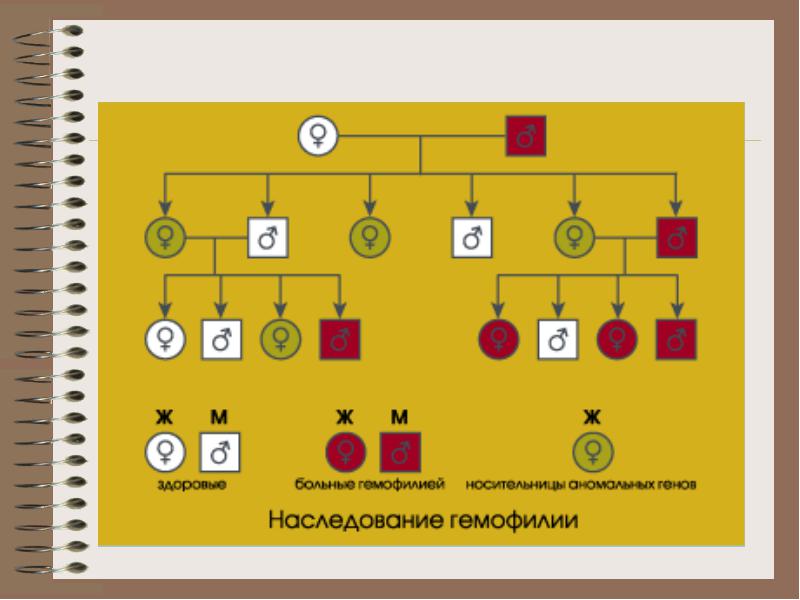
Гемофилия
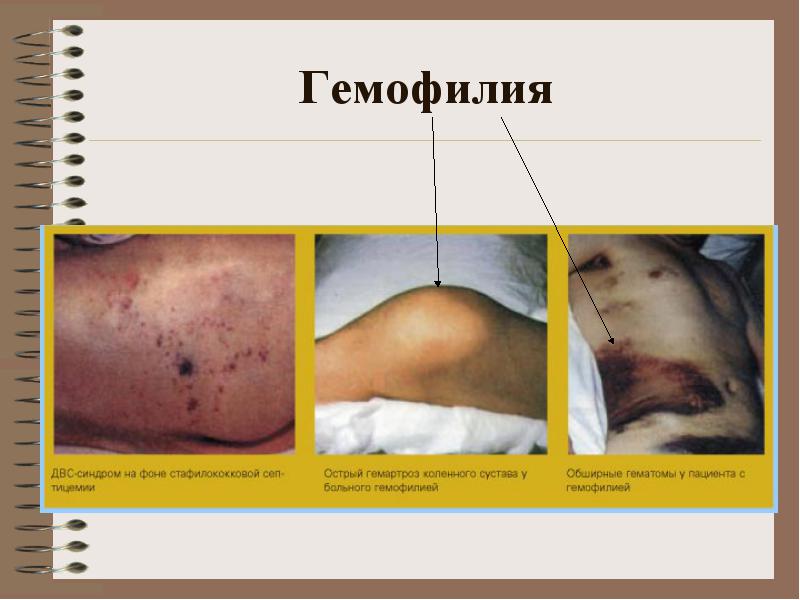
Гемофилия
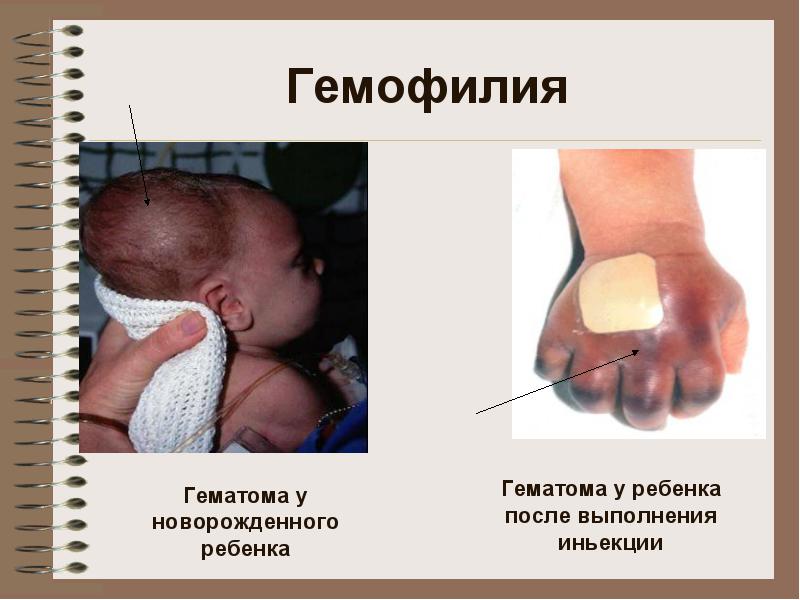
Гемофилия В
Этиология – аномалия гена в Х-хромосоме, который контролирует синтез ф. 9
Болеют – мужчины (46, XhY)
- женщины (46, XhXh), (45, Xh O)
*** Виды
- Гемофилия В+ (антигенположительная форма – синтезируется аномальный ф. 9 )
- Гемофилия В- (антигенотрицательная форма – не синтезируется ф. 9 )
Клиника: кровоизлияния в большие суставы, гематомы (подкожные, внутримышечные), сильные и длительные посттравматические кровотечения. Возможны кровоизлия-ния в органы брюшной полости, желудочно-кишечные кровотечения

ПРИОБРЕТЕННАЯ КОАГУЛОПАТИЯ
Особенность – полидефицитная
Етиология
1. Иммунная ингибиция прокоагулянтов (резус конфликт)
2. Дефицит вит. К–зависимых факторов коагуляции (7, 10, 9, 2)
а) нарушение синтеза в кишечнике (дисбактериоз, понос)
б) нарушение всасывания вит. К (дефицит желчи)
в) тяжелое повреждение печени
3. Передозировка гепарина

ГИПЕРКОАГУЛЯЦИЯ
ПОВЫШЕННАЯ СПОСОБНОСТЬ КРОВИ ОБРАЗОВЫВАТЬ СГУСТКИ В СОСУДАХ
ТРОМБОЗ
ДВС-СИНДРОМ

ДВС-СИНДРОМ (СИНДРОМ ДЕСИМИНОРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ КРОВИ)
КЛАССИФИКАЦИЯ
* По клиническому течению
1) острый (мгновенные формы характеризуются тяжелым течением)
2) хронический
* По распространенности
1) локализированный
2) генерализированный

ЭТИОЛОГИЯ
Инфекции, септические состояния
Шок (при септическом – смертность 100 %)
Хирургические вмешательства, ожеги
Все терминальные состояния, остановка сердца
Острый внутрисосудистый гемолиз
Акушерская патология (20-25 %)
Гемобластозы (при о. лейкозе – 33-45 %)
Деструктивные процессы в паренхиматозных органах
Аллергические реакции

Стадии ДВС-синдрома
1) Гиперкоагуляция (образование множественных тромбов вследствие активации системы коагуляции)
2) Коагулопатия потребления (истощение системы каогулянтов, чрезмерное использование тромбо-цитов для образования тромбов)
3) Гипокоагуляция (понижение активности коагу-лянтов, активация антикоагулянтов, активация фибринолиза)
4) Завершение (выздоровление, осложнения, смерть)

Патогенез ДВС-синдрома
1) Гипертромбинемия (тромбопластин поступает в кровь в большом количестве из поврежденных тканей и способ-ствует образованию тромбина). При инфекциях активированные моноциты-макрофаги синтези-руют собственные коагулянти (ф.7, ф.10, ф.9, ф.2)

Патогенез ДВС-синдрома
2) Массивная агрегация тромбоцитов (вызывает развитие тромбоцитопе-нии потребления и осложняется геморрагиями)
3) Травматизация и гемолиз эритро-цитов (при этом выделяется много АДФ, что усиливает адгезию и агрегацию тромбоцитов)

Патогенез ДВС-синдрома
4) “Гуморальный протеазный взрыв” (при активации прокоагулянтов, ан-тикоагулянтов, фибринолитиков, белков калликреин-кининовой си-стемы, системы комплемента в крови накапливается много продуктов бел-кового распада, которые являются очень токсическими и повреждают сосуды и ткани)

Патогенез ДВС-синдрома
5) Истощение системы фибрино-лиза
(способствует тромбообразованию)
6) Истощение факторов свертывания крови
(вызывают развитие геморрагий)

Клиника ДВС-синдрома
1. Гемокоагуляционный шок
причина
* нарушения микроциркуляции (вызывают развитие гипоксии тканей)
* накопление токсических продуктов протеолиза
проявления
* понижение артериального давления
* понижение центрального венозного давления
* кровотечения (провоцируют геморрагический шок)

Клиника ДВС-синдрома
2. Нарушения гемостаза
а) гиперкоагуляция
Главное проявление – тромбоз
Кровь сворачивается в пробирке
б) гипокоагуляция
Главное проявление – кровотечения
(одновременно истощается система фибринолиза)

Клиника ДВС-синдрома
3. Тромбоцитопения
Возникает вследствие образова-ния большого количества тромбов в сосудах (тромбоцитопения потребления)

У девочки после перенесенной ангины появилась петехиальная сыпь на коже конечностей и туловище. Объективно: количество тромбоцитов 80 Г/л, антитромбоцитарные антитела. Аллергические реакции которого типа (по Кумбсу и Джеллу) лежат в основе этого заболевания?
@ II типа (гуморальные цитотоксические)
I типа (анафилактические)
III типа (иммунокомплексные)
V типа (стимулирующие)
У больного нарушена адгезия тромбоцитов к коллагену и наблюдаются кровотечения из мелких сосудов. Нарушение какого звена гемостаза можно допустить у больного?
@ Сосудисто–тромбоцитарного
Коагуляционного, И фазы
Коагуляционного, ІІІ фазы
Фибринолиза
Коагуляционного, ІІ фазы
У больного диагностирована тромбоцитопения. Какие клинические проявления типичны для нарушений тромбоцитарно–сосудистого гемостаза?
@ Петехии, экхимозы (кровоподтеки)
Гемартрозы
Гематомы
Уменьшение времени кровотечения
Увеличение времени свертывания крови
У больной при обследовании обнаружили тромбоцитопатию. Укажите, какое изменение играет важную роль в патогенезе тромбоцитопатий?
@ Продукция патологических тромбоцитов костным мозгом
Снижение активности антикоагулянтов
Гиперактивация тромбоцитопоэза
Повышение концентрации в крови прокоагулянтов
Угнетение фибринолиза.
Перед проведением оперативного вмешательства обнаружено, что у человека время кровотечения увеличено до 10 мин. Дефицит каких форменных элементов в составе крови может быть причиной таких изменений?
@ Тромбоцитов
Эритроцитов
Моноцитов
Лимфоцитов
Лейкоцитов
У пациентки длительное употребление аспирина вызывало кровоизлияния. Объективно: тромбоцитопения с нарушением функциональной активности тромбоцитов. Тромбоцитопатия в данном случае вызвана угнетением активности:
@ Циклооксигеназы
Цитохромоксидазы
Липооксигеназы
Супероксиддисмутазы
Фосфолипазы А 2 .
У больного диагностировано уменьшение продукции эндотелием сосудов фактора Виллебранда. Какое нарушение сосудисто–тромбоцитарного гемостаза наблюдается при этом?
@ Нарушение адгезии тромбоцитов
Нарушение агрегации тромбоцитов
Гиперкоагуляция
Нарушение полимеризации фибрина
Усиление фибринолиз
У ребенка с геморрагической сыпью, которая возникла после ОРВИ, диагностирован геморрагический васкулит (болезнь Шенляйн – Геноха). Аллергические реакции которого типа (по Кумбсу и Джеллу) лежат в основе этого заболевания?
@ III типа (иммунокомплексные)
I типа (анафилактические)
II типа (гуморальные цитотоксические)
IV типа (клеточные цитотоксические)
V типа (стимулирующие)
У ребенка с геморрагической сыпью, которая возникла после ОРВИ, диагностирован геморрагический васкулит (болезнь Шенляйн – Геноха). Что вызывает нарушение гемостаза при этом заболевании?
@ Повреждение сосудистой стенки
Наследственный дефект соединительной ткани сосудистой стенки
Наследственный дефицит антикоагулянтов
Угнетение фибринолиза
Наследственный дефицит прокоагулянтов
У женщины, которая страдает на желчекаменную болезнь обнаружен геморрагический синдром, обусловленный дефицитом витамина К. Какой из перечисленных факторов является неполноценным при гиповитаминозе К?
@ Стюарта – Прауэра (ф. X)
Фактор Виллебранда
Фибринстабилизирующий (ф. XIII)
Фибриноген (ф. I)
У пациента при обследовании была обнаружена тромбофилия (ускорение процесса свертывания крови). Что могло стать причиной нарушения?
@ Дефицит ингибиторов протеолитических ферментов
Повышение концентрации простациклина
Снижение концентрации тромбину в крови
Повышение концентрации гепарину в крови
Повышение концентрации фибринолизину в крови
У больного с заболеванием печени обнаружено снижение содержания протромбина в крови. Это приведет к нарушению прежде всего:
@ Второй фазы коагуляционного гемостаза
Фибринолиза
Третьей фазы коагуляционного гемостаза
Сосудисто–тромбоцитарного гемостаза
Первой фазы коагуляционного гемостаза
У мальчика 7 лет, после падения с велосипеда возник гемартроз коленного сустава. Введение криопреципитата и откачивание крови из сустава привело к значительному улучшению состояния ребенка. О каком заболевании следует думать?
@ Гемофилия А
Геморрагический васкулит.
Тромбоцитопатия
Тромбоцитопения
Ревматоидный артрит
У мальчика с выраженным геморрагическим синдромом в плазме крови отсутствующей антигемофильний глобулин А (фактор VIII). Какая фаза гемостаза первично нарушена у этого ребенка?
@ Внутренний путь активации протромбиназы
Ретракция кровяного сгустка
Внешний путь активации протромбиназы
Мальчик страдает гемофилией. Какими клиническими признаками проявляются нарушение коагуляционное гемостазу?
Петехиальными кровоизлияниями
Микрогематурией
Экхимозами (кровоподтеками)
Нарушением остроты зрения
@ Гематомами, длительными кровотечениями
У пострадавшего в результате ДТП через некоторое время после перенесенной политравмы развился синдром диссеминированного внутрисосудистого свертывания крови (ДВС). Какой фактор был инициатором этого осложнения?
@ Тканевой тромбопластин (ф. III)
Фибриноген (ф. I)
Антигемофильный глобулин А (ф. VIII)
У больного острым панкреатитом развился синдром диссеминированного внутрисосудистого свертывания крови (ДВС). Какое вещество было инициатором этого осложнения?
@ Трипсин
Фибриноген (ф. I)
Антигемофильный глобулин А (ф. VIII)
Фактор Стюарта – Прауэра (ф. X)
Антигемофильный глобулин В (ф. IX)
В больного с политравмой и острой почечной недостаточностью, состояние осложнилось внутренним кровотечением. Что является главным звеном патогенеза этой стадии ДВС – синдрома?
@ Потребление факторов свертывания крови
Выброс лейкоцитов из депо
Тромбоцитоз
Активация протромбиназы
Угнетение фибринолиза
У больного с ожоговой болезнью как осложнение развился ДВС – синдром. Какую стадию ДВС – синдрома можно заподозрить, если известно, что кровь больного свертывается меньше чем за 3 мин.?
@ Гиперкоагуляции
Гипокоагуляции
Восстановление
Латентную
Терминальную
У больного с травмой развился ДВС – синдром. Какие изменения гемостаза наблюдаются во ІІ фазу ДВС – синдрома?
@ Гипокоагуляция
Гиперкоагуляция.
Фибринолиз
Тромбоцитопения
Тромбоцитопатия
У пациента после переливания несовместимой крови возник ДВС – синдром. Что является главным звеном в патогенезе этого осложнения при гемолизе?
@ Поступление в кровь внутриклеточных протеаз
Накопление билирубина в крови
Избыток в крови тромбопластина
Избыток в крови протромбина
Увеличение содержания плазминогена
У больного на фоне хронической почечной недостаточности развился ДВС–синдром. При обследовании обнаружено увеличение времени свертывания крови, тромбоцитопения, повышение уровня фибрин–мономерных комплексов и продуктов деградации фибрина. О какой стадии ДВС–синдрома следует думать?
@ Гипокоагуляции
Гиперкоагуляции
Восстановление
Латентную
Нестабильную
У больного на хронический лимфолейкозом возникли геморрагии в результате развития ДВС – синдрома. Какие изменения показателей периферической крови будут наблюдаться при этом?
@ Гипокоагуляция, тромбоцитопения
Эритроцитоз, повышение вязкости крови
Тромбоцитоз, уменьшение времени свертывания крови
Гиперкоагуляция, увеличение агрегации тромбоцитов
Повышена активность прокоагулянтов
У женщины с сепсисом появилось петехиальные кровоизлияния, в крови уменьшилось содержание тромбоцитов и фибриногена, появились продукты деградации фибрина. Что может быть причиной отмеченных изменений?
@ ДВС синдром
Лейкопения
Лимфопения
Тромбоцитоз


